
Abstract
Rapid and effective drug discovery for neurodegenerative disease is currently impeded by an inability to source primary neural cells for high-throughput and phenotypic screens. This limitation can be addressed through the use of pluripotent stem cells (PSCs), which can be derived from patient-specific samples and differentiated to neural cells for use in identifying novel compounds for the treatment of neurodegenerative diseases. We have developed an efficient protocol to culture pure populations of neurons, as confirmed by gene expression analysis, in the 96-well format necessary for screens. These differentiated neurons were subjected to viability assays to illustrate their potential in future high-throughput screens. We have also shown that organelles such as nuclei and mitochondria could be live-labeled and visualized through fluorescence, suggesting that we should be able to monitor subcellular phenotypic changes. Neurons derived from a green fluorescent protein–expressing reporter line of PSCs were live-imaged to assess markers of neuronal maturation such as neurite length and co-cultured with astrocytes to demonstrate further maturation. These studies confirm that PSC-derived neurons can be used effectively in viability and functional assays and pave the way for high-throughput screens on neurons derived from patients with neurodegenerative disorders.
Introduction
Efficient drug discovery for central nervous system (CNS) disorders requires high-throughput screening with cultured cells that faithfully recapitulate the human disease. Using high-throughput screens, a large number of compounds could be tested on a primary cell line of neural cells to investigate parameters such as cell viability and phenotypic changes. As of yet, no efficient and reproducible in vitro system by which to conduct these screens has been developed, due in part to the difficulty associated with isolating and testing primary neural cells. Historically, screens for CNS diseases have been conducted on immortalized fibroblast lines, nervous system tumor cell lines, and immortalized neuronal progenitor cell lines, rather than on CNS neurons or astrocytes.1–3 Although all of these lines grow readily in culture, they are generally unstable and often fail to retain critical features of the endogenous neural cells that are required for an effective screen. 4 As such, screening efforts are significantly hampered by an inability to perform experiments in an appropriate cellular context.
Attempts to bypass the limitations presented by screening alternative cell types have included sourcing primary cells from tumor biopsies and cadaveric samples. The advantage of sourcing from these samples lies in the population of adult stem cells present in the nervous tissue, which remain undifferentiated in the body after development and can be grown in culture to obtain neural cells. However, the adult stem cell population is scarce, limited in its potential for self-renewal, and undergoes senescence after a certain number of passages, making it difficult to generate the large number of cells required for screens. 5 Fetal tissue, which is abundant in astrocyte and oligodendrocyte precursors, obviates issues of self-renewal and senescence because these cells possess high proliferative potential. 6 In addition, efforts to immortalize primary cell lines have met with limited success, as many of these cells will transform, develop karyotypic abnormalities, or lose the ability to differentiate.7–9
Breakthroughs in stem cell biology have identified pluripotent stem cells (PSCs), including human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs), as a source by which to develop the specific neural cell types required for high-throughput screening for CNS disorders.10,11 PSCs, unlike adult stem cells, can be propagated indefinitely, have the ability to generate cells of any germ layer, and can be expanded to study fundamental mechanisms of development and investigate potential restoration of tissue damaged by disease or injury. 12 The use of iPSCs is more varied in scope, as it is now possible to isolate and reprogram somatic cells from patients to generate iPSC lines that carry a distinct mutation, a defined genetic background, and a functional history.13,14
Several groups have developed methods to generate CNS cell derivatives from pluripotent stem cells.15–17 Despite these successes, routine screening will require improvements regarding variability within the differentiation process, cell loss during differentiation, the heterogeneity of the resulting cell population, and contamination by other cell types. Additional issues include the long amount of time needed to create neural cells and the relative immaturity of neurons derived using the current differentiation protocols.
In this article, we describe a culture system that uses PSC-derived neural stem cells (NSCs), which can be differentiated into neurons in 96-well plates and tested for high-throughput and high-content (phenotypic) screening purposes. We have previously shown that these NSCs can be obtained from PSCs as a near homogenous population without sorting and can be propagated and cryopreserved without losing their differentiation ability or proliferative capacity.17,18 Mature NSC-derived cortical neurons can be generated within 14 days using a culture system that can easily be adapted to generate neuronal subtypes including dopaminergic neurons and motor neurons. This system is compatible with NSCs derived from various PSCs, including disease line iPSCs and engineered green fluorescent protein (GFP)–expressing hESCs. This article highlights a cell culture platform that can generate a pure population of human neurons from multiple NSC lines and the utility of these neurons in a variety of screening applications.
Materials and Methods
Neural Stem Cell Culture
NSC lines were generated from the episomally derived NCRM-1 iPSC line, patient-derived Niemann-Pick type C (NP-C) iPSC line, patient-derived amyotropic lateral sclerosis (ALS) iPSC line, H14 hESC line, and an engineered H9 (iC23-GFP) hESC line with a ubiquitously expressed GFP-reporter 19 via an embryoid body (EB) intermediate method 17 by XCell Sciences (Novato, CA) and maintained on culture dishes coated with Geltrex (Life Technologies, Carlsbad, CA). Some NSC lines were also made by a direct non-EB–mediated method. 18 The NSC medium was composed of neurobasal media, with L-glutamine (2 mM) and 1× NEAA, 1× B27 supplement, and 10 ng/mL of basic fibroblast growth factor (bFGF; all from Invitrogen, Carlsbad, CA).
Neural Differentiation
For neuronal differentiation, NSCs were split into a 96-well plate coated with 0.002% poly-L-ornithine (Sigma, St. Louis, MO) and 10 µg/mL laminin (Life Technologies) at 7500 to 10,000 cells/cm2, and neuronal differentiation medium was added 24 h after plating. The differentiation medium contained DMEM/F12 with GlutaMax, 1.8% bovine serum albumin (BSA), 1× StemPro hESC supplement (all from Life Technologies), 10 ng/mL brain-derived neurotrophic factor (BDNF) and glial cell line–derived neurotrophic factor (GDNF; R&D Systems, Minneapolis, MN), and cells received fresh medium and growth factors every other day. GABA-ergic forebrain neurons were generated by supplementing the described culture medium with 20 ng/mL Activin (R&D Systems). Astrocyte differentiation and dopaminergic neuron differentiation were conducted as previously described.20,21
Motor Neuron Differentiation
Motor neurons were created in two phases. In the initial neural induction and regionalization phase, neuronal differentiation medium was supplemented with 8 ng/mL bFGF (Life Technologies), 500 µM retinoic acid (RA; Sigma), 200 ng/mL Sonic hedgehog (SHH; R&D Systems), and 20 ng/mL Activin (R&D Systems) for the initial 12 days of the differentiation process. The culture medium was changed every other day, and RA was supplemented daily. During this initial period, cells were passaged 2 to 3 times. After 12 days, the motor neuron maturation phase was initiated by completely withdrawing RA and SHH, reducing the concentration of bFGF to 4 ng/mL, and maintaining 20 ng/mL Activin. In addition, the medium was supplemented with 10 ng/mL BDNF and GDNF (both R&D Systems). The complete medium was changed every alternate day in the maturation phase, and the culture was passaged once at approximately day 22 to 24 after the initiation of the differentiation protocol.
Immunocytochemistry
Immnocytochemistry and staining were performed as described previously. 20 Briefly, cells were fixed with 4% paraformaldehyde for 15 min at room temperature. Fixed cells were washed with phosphate-buffered saline (PBS) twice and blocked using a buffer containing 10% goat serum, 4% BSA, and 0.1% Triton X-100 for 1 h at room temperature. Primary antibody was diluted in antibody buffer containing 8% goat serum, 4% BSA, and 0.1% Triton X-100, and incubation was performed at 4 °C overnight. Appropriate secondary antibodies were used for single and double labeling. The primary antibodies used here are as follows: anti–glial fibrillary acidic protein (GFAP) antibody (DAKO, Glostrup, Denmark), anti-βIII tubulin clone SDL 3D10 (Sigma), anti-Nestin (BD Transduction Laboratories, Franklin Lakes, NJ), anti-GABA (Sigma), anti-HB9 (DSHB, Iowa City, IA), anti-βIII tubulin (Millipore, Billerica, MA), anti-MAP2 (Millipore), and anti-TH (Cell Signaling Technology, Beverly, MA). Secondary antibodies used were Alexa Fluor 488 Goat Anti-Rabbit, Alexa Fluor 594 Goat Anti-Rabbit, Alexa Flour 488 Goat Anti-Mouse, and Alexa Flour 594 Goat Anti-Mouse (Life Technologies). Hoechst 33342 (Molecular Probe H3570) at 1:1000 dilution was used for nuclei staining. Images were captured using a Leica fluorescence microscope.
Gene Expression Analysis
Gene expression analysis was performed on three iPSC-derived NSC samples, three neuron samples derived from those NSCs at day 14, and an iPSC-derived astrocyte line. Samples were collected on different days and processed together. Total RNA was isolated and prepared using the RNeasy Mini kit according to the manufacturer’s instructions (Qiagen, Valencia, CA). RNA samples were hybridized to Illumina Human HT-12 BeadChip by Qiagen (Frederick, MD). Data processing was performed using the algorithms included with the Gene Expression module of the Illumina BeadStudio software with the background method used for normalization. The dendrogram was constructed by global array clustering of genes across all the experimental samples using the complete linkage method and measuring the Euclidian distance. The maximum expression value of gene for the probe set was used as the expression value of the gene. Data were exported to Microsoft Excel for generation of gene lists for assessing the presence or absence of cell-specific markers.
Cell Viability Assays
NCRM-1 NSCs were plated at 10,000 cells/well in a 96-well plate and differentiated as described above. At day 7, neuronal media were changed and cells were reincubated at 37 °C for 1 h prior to addition of the toxic compounds hydrogen peroxide (H2O2) or 3-nitropropionic acid (3-NP). After the 24 h incubation period, the cell viability was determined in each well by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. Assays were performed on three plates with eight replicates per treatment per plate. Statistical significance was assessed by one-way analysis of variance, with groupwise comparisons made with Tukey’s multiple comparison test.
Organelle Targeting
NCRM-1 NSCs were plated at 10,000 cells/well in a 96-well plate and underwent neuronal differentiation as described above. At day 7, mitochondria were targeted using MitoTracker Green FM (Life Technologies) at 50 nM as per the manufacturer’s instructions. Cells were incubated in MitoTracker Green for 30 min at 37 °C. Media were changed after 30 min, and cells were imaged on a Leica microscope at 40×. Neuronal nuclei were targeted using BacMam CellLights Reagents in which GFP contains nuclear targeting sequences (Life Technologies). Neurons were infected with 50 baculoviral particles per cell and imaged with a Leica fluorescence microscope 48 h later.
Time-Lapse Imaging of Neuronal Differentiation
iC23-GFP NSCs were plated in a 96-well format and set for neuronal differentiation as previously described. The plate was imaged using an Incucyte Zoom (Essen Bioscience, Ann Arbor, MI) every 2 h for 14 days. Cells were imaged under phase and fluorescence. Analysis was performed using Incucyte’s NeuroTrack software.
Neuron/Astrocyte Co-culture
Co-culture experiments were performed as described previously. 20 Astrocytes were generated from H14 hESC-derived NSCs, and neurons were derived from iC23-GFP NSCs using our previously published protocol. 21 At day 30, differentiated astrocytes were plated on a layer of neurons that had been differentiated for 21 days (40,000 astrocytes per 1,000,000 neurons). Cells were kept in astrocyte differentiation medium following co-culture for the next 10 days. On day 10, the medium was removed, and cells were washed with PBS, fixed, and stained with SYNAPSIN-1 (1:500; Abcam, Cambridge, MA) and Tuj1 (1:1000; Sigma-Aldrich) antibodies according to the immunostaining procedure described previously. Randomly selected neurons were used to quantify synapse formation. For quantification, neuron selection was based on the criteria that each selected neuron must have an obvious cell body that is separated and distant from other neurons by at least three cell bodies. On the selected neurons, yellow puncta (resulting from co-localization of GFP and SYNAPSIN though image overlay) were identified along a single axon and counted by analyzing fluorescence images using Photoshop. Forty-five randomly selected neurons from three independent experiments were counted blindly by three different individuals. Fold difference in number of yellow puncta by ESC-derived neurons in the presence or absence of astrocytes was calculated by the mean number of puncta counted. Statistical analyses were performed using Student t test with two-tailed distribution and assuming equal variance.
Results
Simplified Culture System to Generate a Pure Population of Neurons from Healthy and Patient-Specific Lines
In this article, we describe a simplified culture system to generate pure populations of neurons from PSC-derived NSCs. PSC samples can be obtained from patients or healthy donors and specialized to form a stable NSC stage.
17
NSCs can self-renew and be cultured for multiple passages without reducing expression of NSC markers or losing their differentiation ability.
18
These cells can also be directly differentiated to multiple neural cells including astrocytes and neurons through manipulation of cell culture conditions, namely, the addition of specific growth factors, in a 96-well format (
Fig. 1
). The starting population of NSCs expresses the NSC marker Nestin (
Fig. 1A
). Nearly all NSC-derived neurons express MAP2 after 14 days of culture in neuronal differentiation medium (
Fig. 1B
). By supplementing the base media with growth factors and compounds that establish positional identity and maturation, one can enrich for specific subtypes of neurons, including HB9+ motoneurons (
Fig. 1C
), and, as we have previously shown,
22
TH+ dopaminergic neurons (
Fig. 1D
). In addition, NSCs have the capacity to differentiate into astrocytes expressing the mature marker GFAP, demonstrating their potential to make both neurons and glia (
Fig. 1E
). This standardized process has been conducted in many different starting cell types, including disease lines and engineered lines, demonstrating high reproducibility (
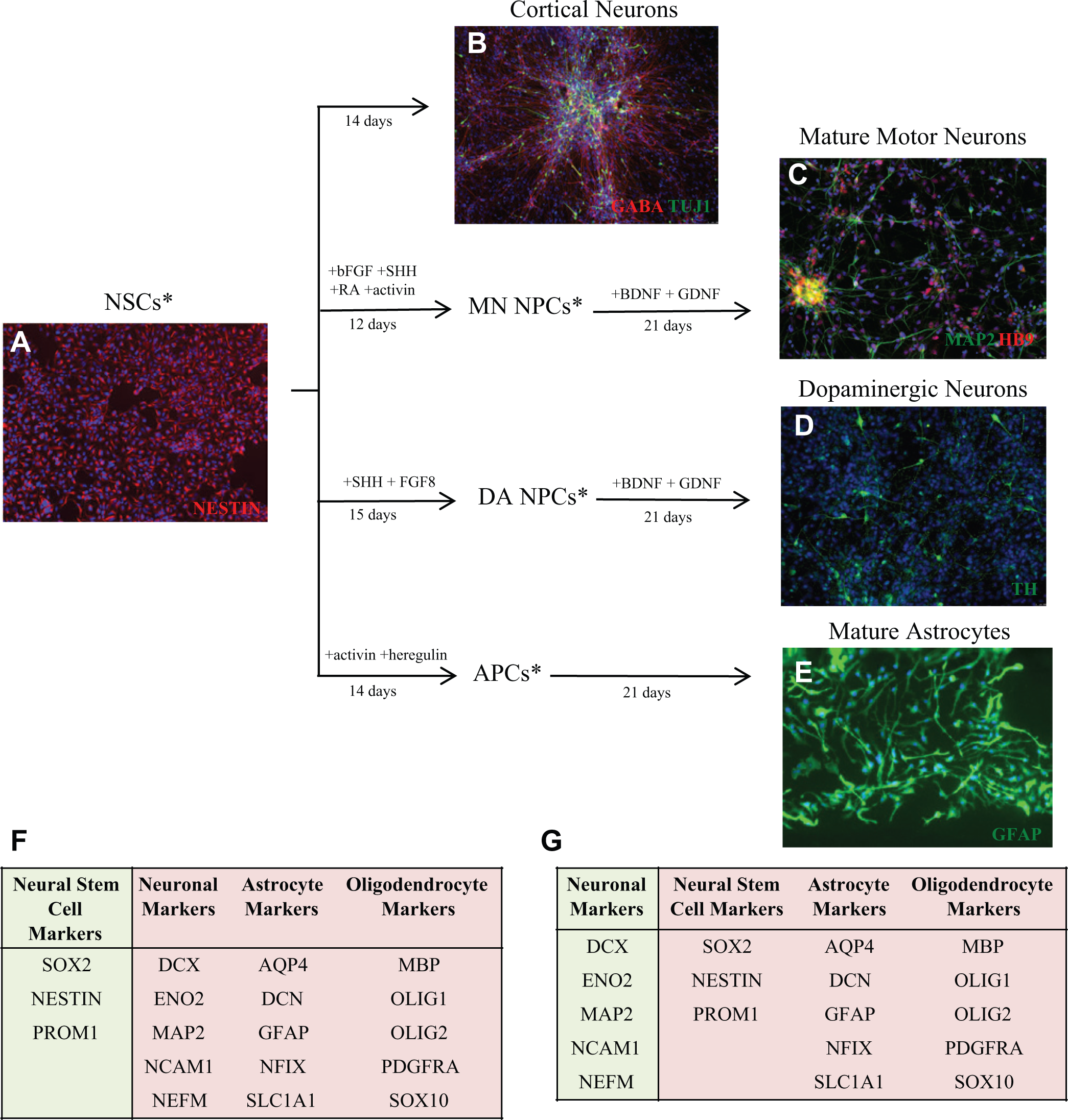
Pluripotent stem cells (PSCs) can be directly differentiated to form pure populations of neurons and astrocytes, which express cell-specific markers. (
Microarray Analysis Confirms Expression of Appropriate NSC and Neuronal Markers in NSCs and Neural Derivatives
Gene expression profiling was conducted on NSCs derived from NCRM-1, NP-C, and ALS iPSC lines, along with neurons differentiated from each of these lines and astrocytes differentiated from an iPSC-derived NSC line. Disease NSC lines were chosen to illustrate the applicability of the described neuronal differentiation method in multiple cell lines, including patient-derived PSCs. A dendrogram analyzing global expression revealed that NSC, neurons, and astrocytes created distinct clusters, with disease line subclusters, indicating that at the global gene expression level of each of these cell types is very similar (
Cells Can Be Grown in a 96-Well Format to Assess Viability
High-throughput assays and phenotypic screens of NSC-derived neurons will require an ability to differentiate these cells in 96-well plates. We demonstrate that iPSC-derived NSCs can be readily differentiated to neurons that present abundant processes as early as 7 days in culture in the required 96-well format ( Fig. 2A ). To assess the potential of performing high-throughput screens on neurons, we performed an MTT viability assay, in which cells were exposed to varying degrees of oxidative stress by treatment with hydrogen peroxide (H2O2) or the mitochondrial stressor 3-NP ( Fig. 2B ). Neuronal cultures were similarly sensitive to both of these oxidative stressors in a concentration-dependent manner. Significant toxicity was observed in each plate of NSCs as concentrations increased from 10 to 100 µM H2O2 and from 3 to 10 mM 3-NP. The data from these assays with NSC-derived neurons were consistent, with standard errors of 5.4% variation from well to well within treatment groups. Plate-to-plate variability was 6.1% across all treatment groups on three replicate plates of NSC-derived neurons. The high reproducibility of these results along with the low variability from well to well and plate to plate suggest that our culture system can provide an appropriate primary cell source for high-throughput screens of neurons.
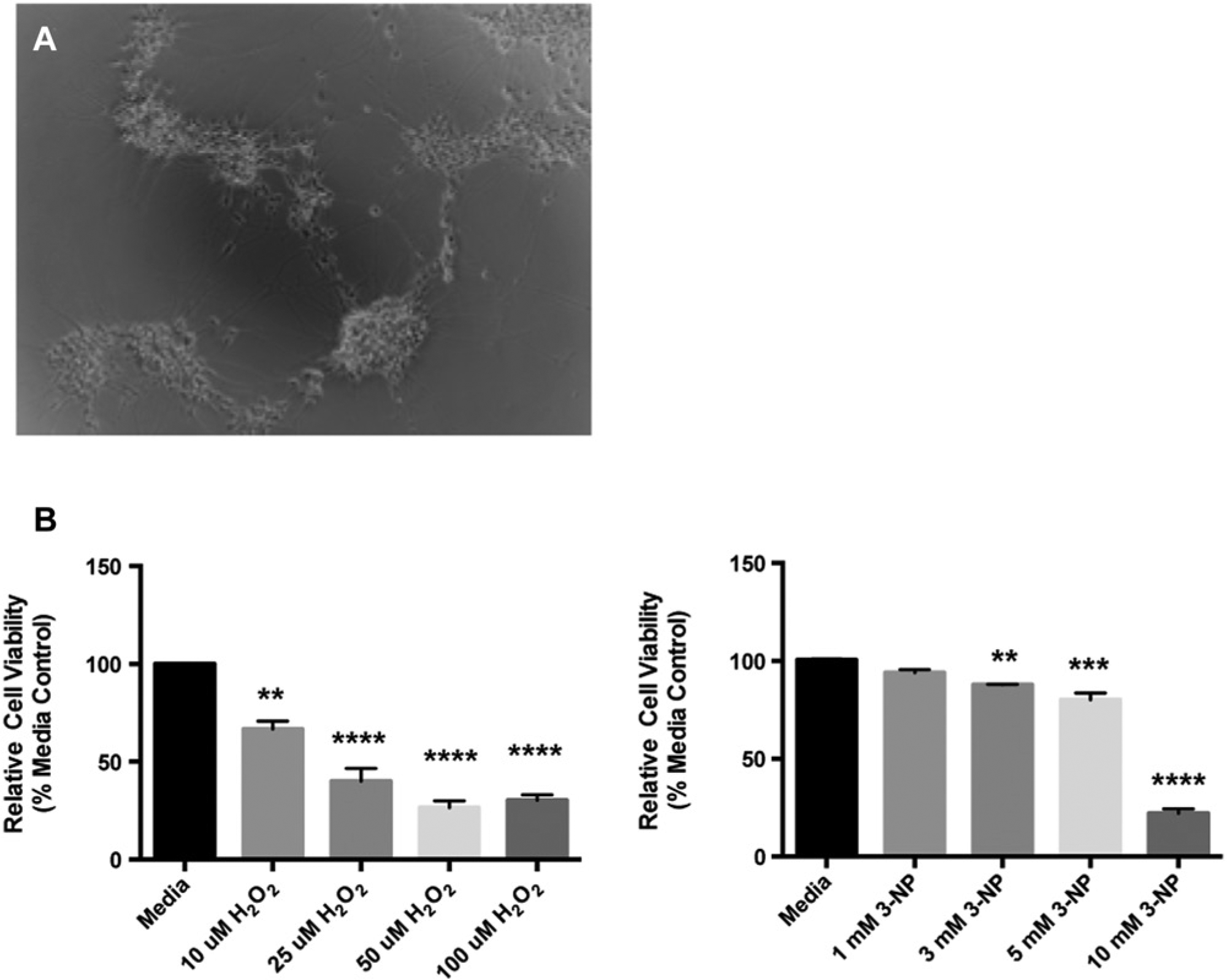
Viability screens can be conducted on differentiated primary neurons in a 96-well format. (
Specific Organelles Can Be Targeted and Monitored within Postmitotic Neurons
High-content screens, or phenotypic screens, monitor phenotypic changes in cells including subcellular changes, changes in overall cell morphology, and changes in gene expression. We sought to label and visualize neuronal organelles in a 96-well format to show that these cells can be used for high-content screens. Mitochondria were targeted using the MitoTracker Green FM mitochondrial live stain, and visualization of the cells displayed distinct fluorescence localized to the mitochondria in all differentiated neurons. Mitochondria were even identified within neurites, suggesting that the movement of these organelles can be tracked using live-imaging techniques ( Fig. 3A ).
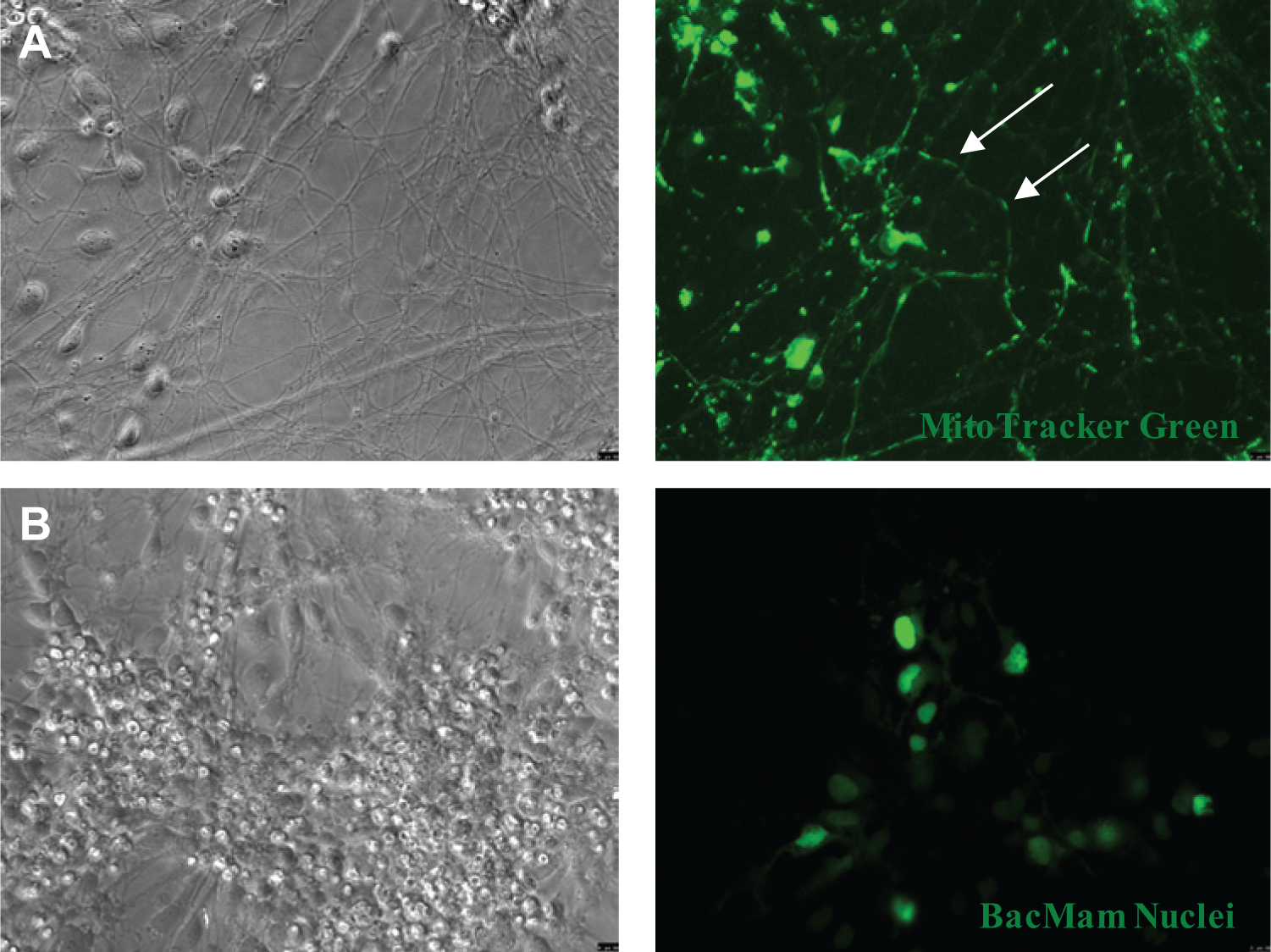
Subcellular organelles can be targeted and identified in postmitotic neurons. (
Postmitotic neurons are extremely sensitive and difficult to transfect. 23 For these reasons, we used BacMam technology to target and label the neuronal nucleus. This technique allows us to target neurons without negative cytotoxic effects. Using BacMam CellLight Reagents, we were able to visualize the neuronal nucleus within 24 h of transfection ( Fig. 3B ). As transfection efficiency was relatively low (<50%), further optimization is necessary. Our ability to successfully target and visualize mitochondria and nuclei in postmitotic neurons suggests that these organelles can be monitored in future phenotypic screens, particularly regarding neurological diseases in which particular organelles are preferentially affected.
Reporter Lines Can Be Generated to Provide Additional Versatility
We have previously developed an hESC line (iC23-GFP) in which a GFP reporter is driven by the chicken β-actin promoter
19
and that could act as a particularly useful phenotypic screening tool. We have generated iC23-GFP NSCs and monitored the neuronal differentiation process in real time using an Incucyte. These NSCs express GFP robustly, with more than 99% of NSCs identified as being GFP+ (
Fig. 4A
). The cells continue to express GFP for up to 14 days as they differentiate in culture (
Fig. 4B
). A compilation of images taken at 2 h intervals during differentiation shows the dynamic changes that occur during a 3 day window selected from days 4 to 7 (
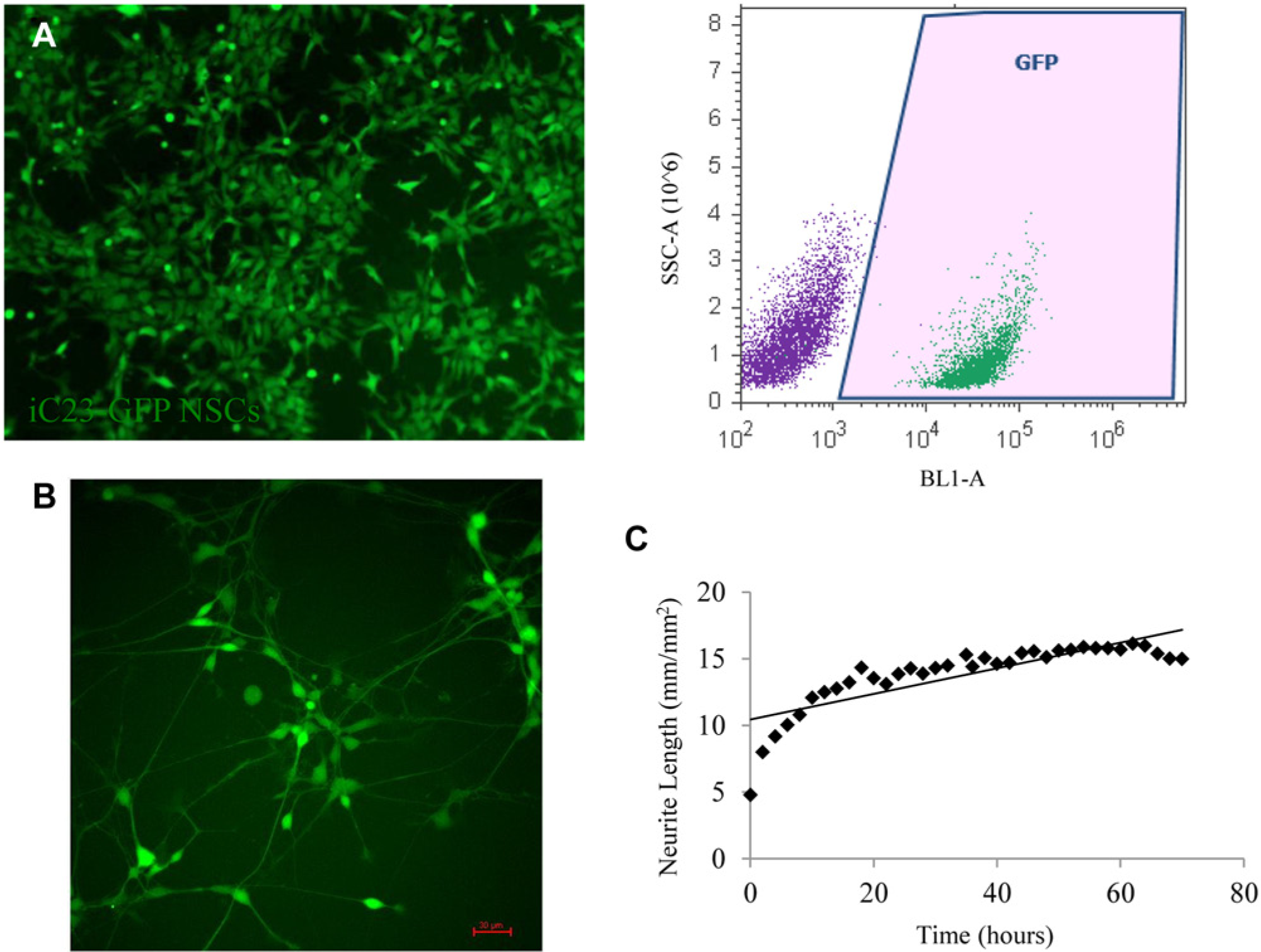
Neuronal differentiation of engineered neural stem cell (NSC) reporter lines can be monitored in real time. (
Neuron/Astrocyte Co-culture Using an Engineered GFP-Expressing Reporter Cell Line
The intimate relationship between neurons and astrocytes in vivo indicates that screens specifically designed for neurodegenerative diseases may benefit from screening in a neuron/astrocyte co-culture system. We have developed a system to co-culture these cells using hESC-derived astrocytes and neurons generated from iC23-GFP NSCs ( Fig. 5 ). Neurons were analyzed by comparing the number of puncta, representative of co-localization of the neuronal marker Tuj1 and the synaptic marker Synapsin1, in neuronal and neuron/astrocyte co-cultures. Neurons displayed a significantly greater number of puncta (~8-fold) in co-cultures as compared with neuronal culture alone. These data indicate that the PSC-derived GFP engineered cell line can be readily cultured in a co-culture model and that neurons grown in this model display increased maturity as indicated by enhanced synapse formation.
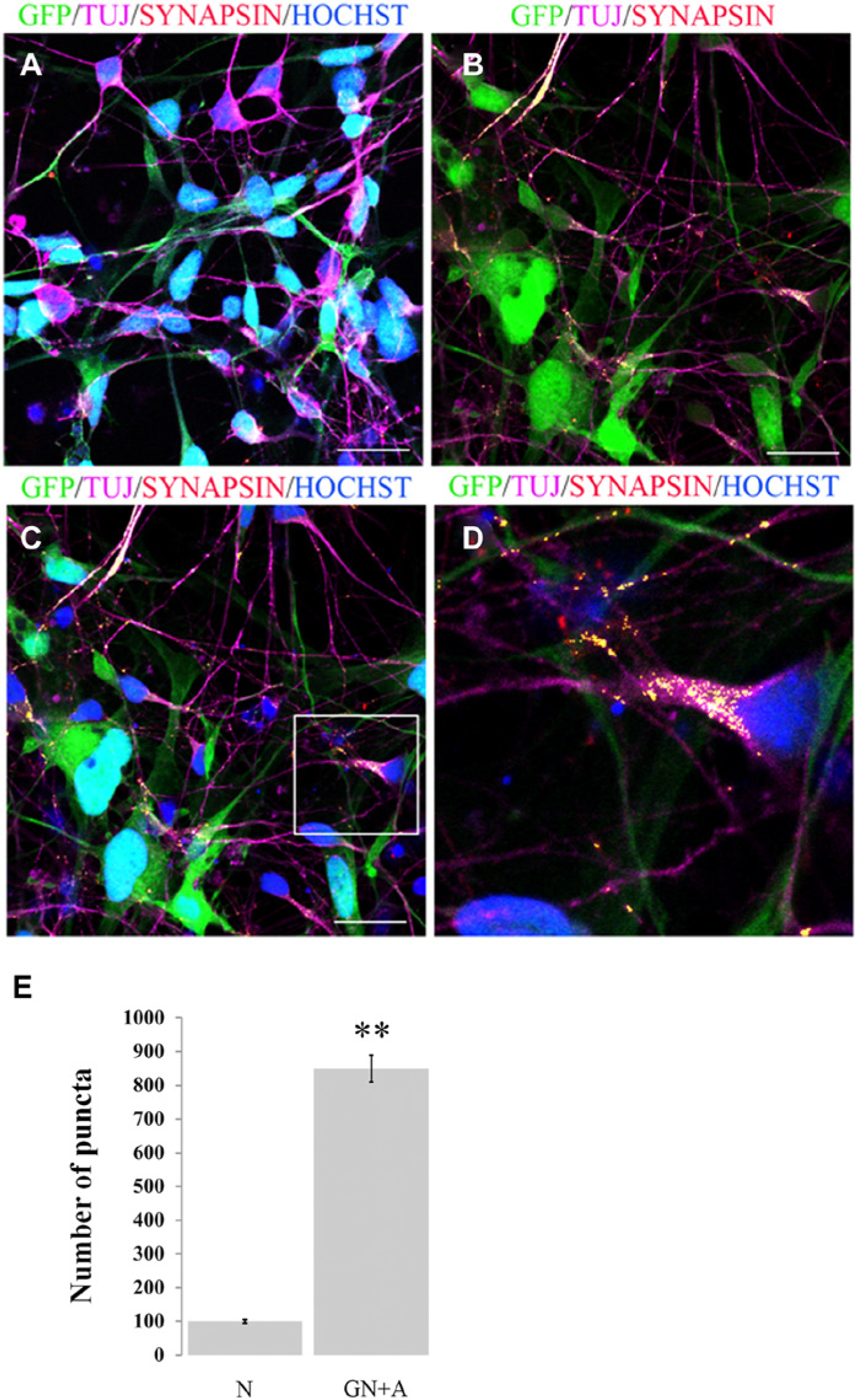
Enhancement of synapse formation by neurons derived from a green fluorescent protein (GFP)–expressing embryonic stem cell (ESC) line H9 (iC23-GFP). (
Discussion
The most critical factor in identifying potent therapeutic agents for neurodegenerative diseases through drug screening will be a source of neural cells that faithfully recapitulate the disease phenotype and can generate a large quantity of pure cell types. We have previously shown that cryopreservable NSCs derived from hESCs or iPSCs meet these criteria and, as such, are an ideal starting point to generate neurons and glia that can be applied to drug discovery platforms.18,19 By using iPSC technology, we can create large panels of cell lines that can then be engineered to create allelic controls. We envision that NSCs could be derived from control, patient, and engineered PSC lines and subsequently screened in a panel using alleleically matched controls. These matched panel sets can be differentiated to neurons and astrocytes, and by placing reporters in post-mitotic cells and performing co-culture, these cells can be used to better mimic the endogenous state. We believe that NSCs or neural cells could be assayed in a variety of high-throughput screens that would facilitate drug discovery and provide mechanistic insights into neural development in both the normal and pathological state ( Fig. 6 ).
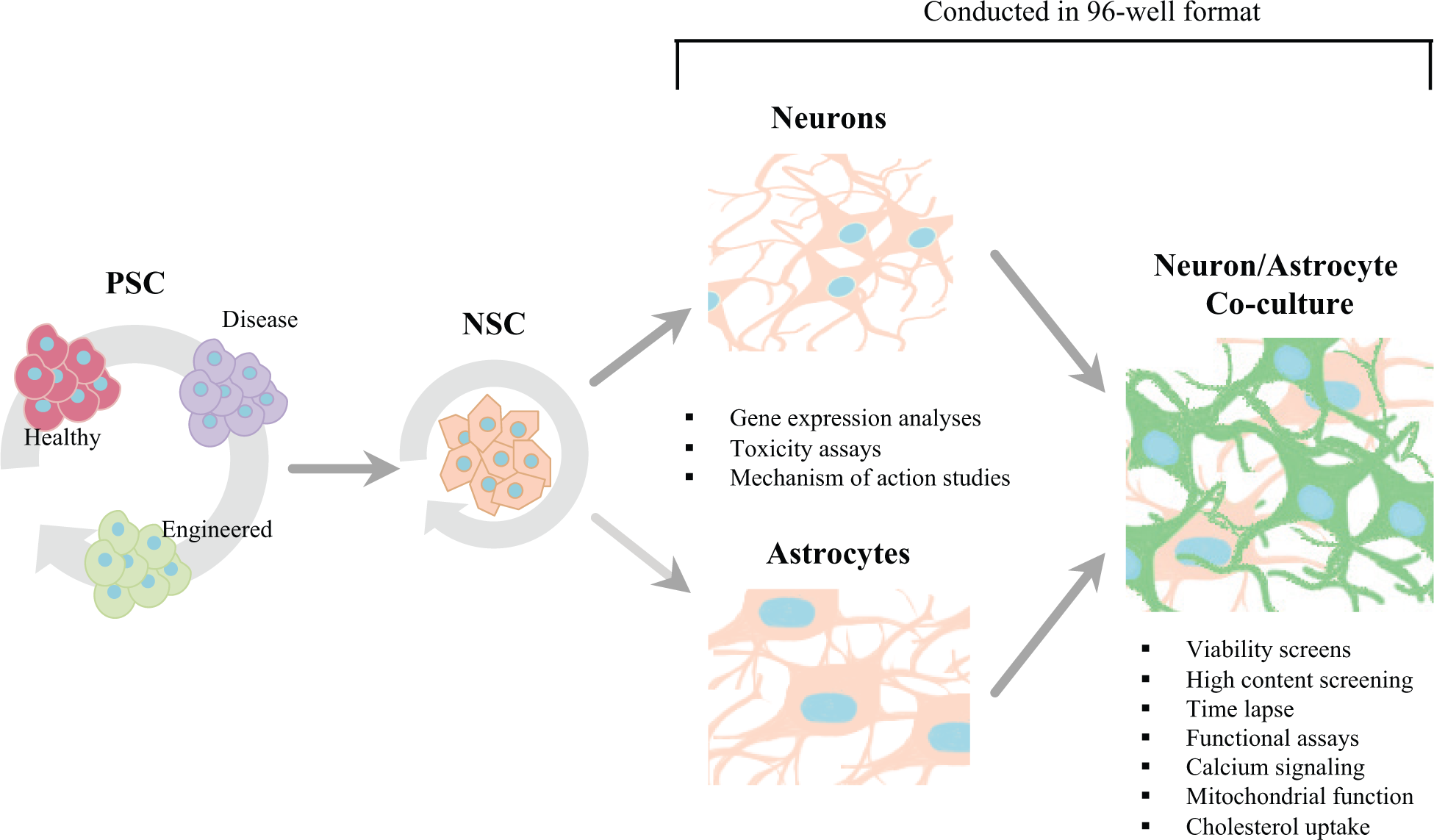
Pluripotent stem cells from healthy, disease, or engineered sources can be bulk cultured and differentiated to self-renewing neural stem cells (NSCs). NSCs can then be further differentiated to neurons or to astrocytes in a 96-well format to perform gene expression analyses, toxicity assays, and mechanism-of-action studies. Neurons and astrocytes can be cultured together or separately for high-throughput viability screens, high-content screening, time lapse, functional assays, calcium signaling, mitochondrial function, or cholesterol uptake assays.
A major finding of this study is the ease with which neurons can be generated and assayed in a 96-well format, which previously had been a major roadblock to the development and conduction of assays for drug discovery for neurodegenerative diseases. Postmitotic adherent neurons with extensive processes do not survive transfers to smaller plates, and the direct plating of PSCs to a 96-well format introduces unacceptable variability. 4 By standardizing this protocol using NSCs as a stable intermediate cell stage, we have created a routine method by which to reproducibly create neurons in a 96-well plate for use in viability assays, visualization of subcellular organelles, and co-culture with astrocytes. Although the differentiated neurons created through this protocol were relatively immature, they expressed key markers at sufficient levels for routine assay needs. We are currently investigating methods by which to generate more mature neurons, including the use of additional growth factors and small molecules, as well as longer-term co-culture with astrocytes. In addition, although we have not applied this protocol in a 384-well format because of the necessity of automated medium changes, we hope to test such a system in the future and believe that neurons can be differentiated using our method in this plate format.
Another critical aspect of performing efficient screens is low well-to-well and plate-to-plate variability. Our cell viability data demonstrate that we can perform a standard cell viability assay on NSC-derived neurons in a 96-well format with low variability and have established an appropriate dynamic range using multiple standard cell toxic agents, thereby suggesting that these neurons are an appropriate cellular source for high-throughput screens on primary cells. These screens may be used to identify compounds that affect factors such as neuronal differentiation and survival, which will necessitate secondary and tertiary screens to analyze dose response and additional effects. Proper characterization of the identified compounds will require testing in an environment that more closely mirrors the endogenous environment, which may include performing screens on co-cultured cells.
Although high-throughput screening focused on cell survival facilitates drug discovery by providing a method to test a large number of compounds rapidly, it does not provide an in-depth analysis of phenotypic changes within cells, including changes within organelles, cell morphology, or gene expression. In targeting specific organelles within postmitotic neurons, we are able to image and analyze changes to these cells at a subcellular level. This has important implications for future screens focused on neurodegenerative disorders. For example, mitochondrial dysfunction in dopaminergic neurons differentiated from patient-specific Parkinson disease iPSCs may be readily assessed through targeting of the mitochondria. 23
A variety of techniques have been developed to visualize neurons. Although stable reporter lines can be generated, this process is time-consuming and was until recently the only method of uniformly labeling postmitotic cells. Labeling using other methodologies such as virus-based transfection methods produces variable results or unacceptable toxicity. 24 We therefore evaluated BacMam as a delivery agent and chose this method to visualize neuronal nuclei based on its versatile and well-characterized system that can be used to target and monitor a variety of organelles within neurons to assess any phenotypic changes. 25 Although we did not test them in this study, BacMam reporters to visualize the endoplasmic reticulum (ER), lysosomes, cytoskeleton, cell surface, and other secretory pathways are also available and could also be used to investigate subcellular dynamics. For example, BacMam reporters for ER can be used to examine ER dysfunction and its relationship to protein folding, which has been implicated in many neurological disorders. 26 These ER and mitochondrial reporters could also be used in conjunction with calcium dyes to study calcium homeostasis in large-scale screening studies, as improper calcium signaling has already been implicated in major neurodegenerative diseases such as Alzheimer disease. 27 As more tools to efficiently target and visualize organelles become commercially available, we envision using these tools in conjunction with other techniques to further investigate cellular phenotype in healthy or disease cells. Our results demonstrate the feasibility of targeting organelles in primary neurons to analyze phenotypic changes in these structures at the subcellular level, which can be adapted to high-content screens.
Phenotypic screens can also be facilitated by the use of engineered cell lines. To demonstrate the potential of engineered PSC lines, we used a line (iC23-GFP) that expresses GFP ubiquitously in PSCs, NSCs, and neurons to track the differentiation of cells under specified conditions. Engineered lines expressing GFP or luciferase in only postmitotic neurons or under lineage-specific promoters would be excellent tools for high-content screens by providing a way to visualize and quantify gene expression in these cells. As previously stated, further characterization of promising compounds identified in primary screens will require conditions that mirror an in vivo cellular context. To mimic the endogenous cellular environment in a 96-well format, we created a co-culture system between the iC23-GFP NSC-derived neurons and unlabeled NSC-derived astrocytes, demonstrating that we could distinguish between each of these cell types in culture and that the neuron/astrocyte co-culture resulted in more mature neurons. Based on our data, we believe that other healthy, disease, and engineered cell lines from multiple PSC sources can be easily adapted to a co-culture system, thereby more faithfully mimicking the endogenous environment that will help in further development of compounds that show promise in primary screens.
We have noted several differences between our cells and currently used cell lines. Primary cells are relatively immature, and although they have an ability to mature over time, they often fail to reach adult levels of gene expression even after months in culture. The cost of growing primary cells is far greater than that of cell lines due to more stringent culture requirements, the need for expensive components, and the prolonged maturation time of these cells. Despite these limitations, primary cells are more similar to endogenous adult cells than cell lines, and we are currently exploring methodology to immortalize a key line at a late stage of differentiation, thereby addressing some of these issues. In summary, we have shown that iPSC- or hESC-derived NSCs are an abundant, reliable source of neurons and astrocytes and that these derivatives can be grown in a variety of cell culture formats and engineered for use in both standard viability screens and high-content screens.
Footnotes
Acknowledgements
The authors thank our colleagues Dr. Kristina Zaal of the NIAMS Light Imaging Section for her assistance on using the Incucyte Zoom for live imaging and Trevor Cerbini for his assistance in performing flow cytometry and analysis. We also thank members of the Rao Lab, particularly Sonia Shah and Ray Funahashi, for their critical reading of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the NIH Common Fund.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.