
Abstract
The mTOR pathway is a critical integrator of nutrient and growth factor signaling. Once activated, mTOR promotes cell growth and proliferation. Several components of the mTOR pathway are frequently deregulated in tumors, leading to constitutive activation of the pathway and thus contribute to uncontrolled cell growth. We performed a high-throughput screen with an isogenic cell line system to identify compounds specifically inhibiting proliferation of PTEN/mTOR-pathway addicted cells. We show here the characterization and mode of action of two such compound classes. One compound class inhibits components of the PTEN/mTOR signaling pathway, such as S6 ribosomal protein phosphorylation, and leads to cyclin D3 downregulation. These compounds are not adenosine triphosphate competitive inhibitors for kinases in the pathway, nor do they require FKBP12 for activity like rapamycin. The other compound class turned out to be a farnesylation inhibitor, blocking the activity of GTPases, as well as an inducer of oxidative stress. Our results demonstrate that an isogenic cell system with few specific mutations in oncogenes and tumor suppressor genes can identify different classes of compounds selectively inhibiting proliferation of PTEN/mTOR pathway–addicted isogenic clones. The identified mechanisms are in line with the known cellular signaling networks activated by the altered oncogenes and suppressor genes in the isogenic system.
Introduction
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase that integrates signals from growth factors, nutrients, and stresses to regulate multiple processes (reviewed in Sarbassov et al. 1 ). Since the PI3K/mTOR pathway is a key regulator of cell growth and proliferation, it is commonly deregulated in many human cancers. 2 Therefore, efforts to identify potent inhibitors targeting this pathway are actively pursued (for review, see Benjamin et al. 3 ).
mTOR exists in two complexes, mTORC1 (comprising raptor) and mTORC2 (comprising rictor), which are differentially regulated and have distinct substrate specificities. Importantly, only mTORC1 is sensitive to inhibition by rapamycin. The mTOR complexes sense the availability of nutrients (e.g., amino acids, glucose) and consolidate this information with growth factor signaling through the PI3K/Akt/TSC pathway. Activated mTORC1 controls the rate of protein synthesis for selected messenger RNAs (mRNAs) via S6K1 (RPS6KB1) and 4E-BP1, whereas mTORC2 modulates cell survival in response to growth factors by phosphorylating Akt/PKB at Ser473 4 and serum/glucocorticoid regulated kinase 1 (SGK1). 5
Accumulating evidence suggests that many of the vulnerabilities of cancer cells reflect states of dependency (“addiction”) that are unique to cancer cells. Such addiction can arise due to a strict dependency on a single activated oncogene, a cell lineage–specific factor, or even to a nononcogene. 6 Although the PI3K/mTOR pathway is often deregulated in cancer, it is not clear what renders cells truly PI3K/mTOR pathway addicted. Noteworthy, mTOR itself is not an oncogene, and activating mutations in mTOR in human cancer are very rare. 7 However, the tumor suppressors PTEN, TSC1/2, and LKB1, which are upstream of mTOR, are often mutated in human cancer and their loss, among other effects, activates mTOR. 8 Inhibition of phosphorylation of S6K1 as well as RPS6 is well accepted as downstream readouts for mTOR pathway inhibition by rapamycin and other mTOR inhibitors. 9 But, as described also by others, inhibition of mTORC1 kinase activity and S6 ribosomal protein phosphorylation does not always correlate with antiproliferative sensitivity of cell lines or tumors in vivo. 10 It therefore appears that selective killing of mTOR pathway–addicted cells requires inhibition of other and/or additional components of the mTOR pathway or of unrelated pathways that are critically important to maintain cellular homeostasis when the PI3K/mTOR pathway is activated. The latter refers to synthetic lethal genetic interactions, classically defined as significantly deleterious or lethal phenotypes resulting from the combination of two or more mutations that do not produce such phenotypes individually. 6 Chemical inhibition of the function of a gene product can be considered equivalent to a genetic loss-of-function mutation. As such, the identification of specific drug targets in cancer cells that are synthetically lethal in combination with mutant genes might allow the specific destruction of cancer cells while leaving normal cells intact. The best example, which has been recently clinically validated, 11 is the finding that inhibition of poly(ADP-ribose) polymerase (PARP) is selectively killing cells with mutations that inactivate the BRCA1/2 genes. 12 Increased rapamycin sensitivity of cells with loss of PTEN or gain of PI3K function is another example. 13
Most synthetic lethality screens are currently performed with short hairpin RNA (shRNA) or small interfering RNA (siRNA) libraries to identify genes that can then serve as molecular targets for drug discovery. Synthetic lethality compound screens have also been performed (for review, see Chan and Giaccia 14 ), mainly searching for synthetic lethal interactions with oncogenic RAS.15–17
To exploit the concept of addiction and synthetic lethality, we used an isogenic cell system comprising Ras-transformed cell lines with additional mutations in the PTEN/PI3K/mTOR pathway to identify compounds that selectively kill PTEN/PI3K/mTOR-pathway addicted cells. The panel includes also non–mTOR-addicted isogenic lines, which were used as specificity controls. Among the different cell lines, we chose clone 6.5, which carries a stop codon in the Pten gene and is rapamycin sensitive for a high-throughput screen (HTS). Interestingly, addition of interleukin (IL)–3 into the cell culture medium efficiently rescued the effect of rapamycin via the activation of the Jak-Stat pathway. 18 The pleiotropic role of PTEN and the convergence of signals from other oncogene and tumor suppressor networks to activate the Akt-mTOR pathway indicate that many points of intervention for pathway inhibition may exist in a cell and that the 6.5 cell line offers an ideal screening tool. Thus, we performed an HTS with clone 6.5 cells in the presence and absence of IL-3. We describe here the identification and characterization of those compounds that showed the highest selectivity for mTOR-addicted cells.
Materials and Methods
Compounds
Compounds were originally obtained from InterBioScreen (Moscow, Russia) (Cpd-1 (8,8-Dimethyl-5-morpholin-4-yl-8,9-dihydro-6H-7-oxa-3-thia-2,4-diaza-cyclopenta[a]naphthalen-1-ylamine) and Cpd-3 (4-Hexyl-1,2,3,4-tetrahydro-cyclopenta [b] quinolin-9-ylideneamine hydrobromide)), ChemDiv (San Diego, CA) (Cpd-2, 1-(4-Ethoxy-phenyl)-2-(4-methoxy-phenyl)-6,7,8,9-tetrahydro-5H-imidazo [1,2-a] azepin-1-ium bromide), ChemBridge (San Diego, CA) (Cpd-4, Diethyl-[2-(4-imino-4H-pyridin-1-yl)-quinazolin-4-yl]-amine), and Tripos (St. Louis, MO) (Cpd-5; rac-[4-(2-Ethyl-phenyl)-1,4,6,7-tetrahydro-imidazo[4,5-c]pyridin-5-yl]-[1-(4-fluoro-phenyl)-5-methyl-1H-pyrazol-4-yl]-methanone) and resynthesized at Actelion Pharmaceuticals (Allschwil, Switzerland). All derivatives where synthesized at Actelion Pharmaceuticals. Rapamycin was purchased from Tocris (Bristol, UK); FTI-277, NSC-663284, Chelerythrine, and NAC (N-acetylcysteine) were from Sigma (St. Louis, MO); and NVP-BEZ-235 and PP242 were from Selleck Chemicals LLC (Houston, TX).
Cells
The isogenic cell line panel was created by the group of Prof. C. Moroni. 18 Murine, PB-3c-15v4 mast cells as well as their derivatives were cultured in IMDM (Sigma) supplemented with 10% fetal calf serum, penicillin, streptomycin, 50 µM β-mercaptoethanol, and, if required, 2 ng/mL murine IL-3 (Sigma). 15V4 cells were transformed by a human bcr-abl (oncogene gift of Dr. T. Klimkait, University of Basel, Switzerland) using the retroviral packaging system of Morita et al. 19 as described. Retroviral supernatants were used to infect 15V4 cells, and as the bcr-abl oncogene transforms cells to IL-3 independence, bcr-abl–expressing cells were selected by growing them in the absence of IL-3. The bcr-abl transduced cells were sensitive to the Abl kinase inhibitor Gleevec (data not shown).
IL-3–independent shPTEN cells were generated from 15V4 cells following knockdown of PTEN via lentiviral shRNA with the target sequence 5′-CGACTTAGA-CTTGACCTATAT-3′ (Sigma TRCN0000028991) and simultaneous introduction of puromycin resistance. Lentiviral particles carrying specific targeting shRNAs and a puromycin-resistance marker in a pLKO1-Puro vector from Sigma were used for infection. Particles were obtained from Sigma at a titer of 1 to 3 × 107 transfection units per milliliter. The 5 × 105 cells were infected in IL-3–containing medium with 5 × 105 transfection units of lentiviral supernatant in the presence of 10 mg/mL polybrene. After 12 h, puromycin selection was applied (2 mg/mL) for at least 36 h. Later, cells were cultured without puromycin and IL-3. Stable FKBP12 knockdown in 6.5 cells was achieved as described above but with the target sequence TGCTTGAA-GATGGAAAGAAAT (Sigma TRCN0000012490) in clone 6.5-shFKBP12-3 and GCCAAACTGATAATCTCCTCA in clone 6.5-SHFKBP12-4 (Sigma TRCN0000012491). The target sequence for the nontargeting control (sh-Control) was 5′-CAACAAGATGAAGAGCACCAA-3′ (Sigma SHC002V).
HTS Assay
An AlamarBlue growth assay in a 384-well format was carried out in an assay volume of 80 µL, with final concentrations of 10 µM compound, 0.5% DMSO, and 10,000 cells/well, according to the following protocol: compound plates were prepared containing 1.2 µL of compound stock solution (2 mM on average) in wells of columns 1 to 22 and DMSO in wells of columns 23 and 24 with the aid of a parallel pipettor (CyBio 384-well liquid handling robot; CyBio AG, Jena, Germany) on the day of assay. Then, 25 µL growth medium without IL-3 was added with a Multidrop dispenser (BioConcept AG, Allschwil, Switzerland). Duplicate assay plates (with differing barcodes) were prepared by transferring 2 × 9 µL from the compound plates to the assay plates with the aid of a parallel pipettor (CyBio 384-well liquid handling robot; CyBio AG). Then, a 70-µL cell suspension in growth medium plus or minus IL-3 was added to the respective assay plates with a Multidrop dispenser (BioConcept AG) for a final concentration of 10,000 cells/well. Assay plates were incubated for 24 h at 37 °C, 5% CO2 in a humidified cell culture incubator. Next, 8 µL AlamarBlue reagent (Biosource/Invitrogen, Basel, Switzerland) was added to all wells with a WellMate dispenser (Thermo Fisher Scientific/Matrix, Hudson, NH). Assay plates were incubated for 8 to 10 h at 37 °C, 5% CO2 in a humidified cell culture incubator. Fluorescence was measured on a Safire II reader (TECAN, Männedorf, Switzerland; excitation = 530–560 nm, emission = 590 nm). The assay was technically performed at BioFocus (Allschwil, Switzerland).
Proliferation Assay with Isogenic Cell Line Panel
Ten thousand cells per well were plated in 135 µL medium (with or without IL-3, depending on the cell line) in 96-well plates in triplicate. Then, 15 µL medium containing a 10× stock solution of inhibitors was added per well, and cells were grown for 48 h with inhibitors. Next, 30 µL Cell Titer Blue (Promega, Madison, WI) was added per well, and plates were incubated for 2 h at 37 °C. Fluorescence was measured with a Safire Multiplate Reader (TECAN) or a SPECTRAMax GeminiXS reader (Molecular Devices, Sunnyvale, CA; excitation = 530–560 nm, emission = 590 nm), and values were normalized to inhibitor-free and cell-free controls. Data are presented as the mean value of the triplicates (± standard deviation). Each experiment was performed two to three times.
For experiments with NAC, cells were seeded in 96-well plates in 135 µL per well ± 10 mM NAC (freshly diluted in water; Sigma) without IL-3.
Proliferation Assays in Human Cancer Cell Lines
Cells were seeded in 96-well plates in 90 µL medium. The following densities were used: WI38 and A498, 1250 cells/well; HCT-116, 2500 cells/well; and A427, CCRF-CEM, RPMI-8226, MOLT4, and HL60, 10,000 cells/well. For all other cell lines, 5000 cells/well were used. The next day, cells were treated with 10 µL 10× concentrated compound solution or 1% DMSO solution (0.1% final DMSO concentration) and incubated for an additional 72 h. Then, 20 µL Cell Titer Blue (Promega) was added, and reading was performed after a 1-h incubation at 37 °C with a SPECTRAMax GeminiXS reader (Molecular Devices; excitation = 560 nm, emission = 590 nm). Each experiment was performed at least twice. In experiments where NAC was used, 10 mM NAC (freshly diluted in water; Sigma) was added to the cells at the time of seeding.
Western Blots
MDA-MB-468 cells were lysed in ice-cold RIPA buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 2 mM EDTA, 1% NP-40, and 0.1% sodium dodecyl sulfate [SDS]), freshly supplemented with protease inhibitor cocktail (PIC; Sigma) and Na2VO3 2-mM final concentration (Sigma). After a 20-min incubation, cells were scraped and cell lysates were transferred into tubes and incubated another 10 min on ice. The 6.5 cells were lysed in ice-cold TNE buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.5 mM EDTA, and 1% Triton X-100 supplemented with protease inhibitors and phosphatase inhibitors [Roche complete protease inhibitor cocktail, 1 mg/mL aprotinin, 1 mg/mL Pefabloc, 1 mM phenylmethylsulfonyl fluoride, 10 mM NaF, 10 mM NaN3, 10 mM NaPPi, 10 mM b-glycerophosphate, and 10 mM p-nitrophenylphosphate; Roche Diagnostics, Rotkreuz, Switzerland). Cell debris was removed by centrifugation for 15 to 30 min at 13,000 g at 4 °C. The protein concentration was determined with the BCA Protein Assay Kit (Thermo Fisher Scientific/Pierce) and equalized among samples. For Western blotting, between 10 and 30 µg protein was loaded per lane. All primary antibodies were from Cell Signaling (Beverly, MA), except the anti–un-prenylated Rap1a antibody, which was from Santa Cruz Biotechnology (Santa Cruz, CA); the anti–cyclin D3 antibody, which was from Becton Dickinson (San Diego, CA); and the anti–actin antibody, which was from Sigma. Each Western blot was repeated at least twice.
In Vitro Kinase Assays
Kinase assays were performed at Invitrogen (Madison, WI) using the Z′-LYTE screening protocol and assay conditions, except for the PI3K assay, in which the Adapta screening protocol and assay conditions were used.
Results
Isogenic Cell System
As described by Nair et al.,
20
the IL-3–addicted v-H-Ras–expressing mouse mast cell line PB-3c-15v4 (called 15v4 in this article) was mutagenized and IL-3–independent clones were isolated (
Fig. 1A
). Many of these IL-3–independent clones were shown to have an activated mTOR pathway.18,21 Several clones had mutations or deletions of the tumor suppressor PTEN, such as clone 6.5. The clone designated D2c harbors a mutation in the tsc2 gene, leading to complete absence of TSC2 protein (
Fig. 1B
). Other clones had mutations in the IL-3 pathway, for example, the clone D5a, which shows constitutive active STAT5 phosphorylation even in the absence of IL-3, mimicking constitutive IL-3 signaling.
18
As expected, all the clones with an activated mTOR pathway were sensitive to rapamycin, whereas D5a, as well as a clone engineered to overexpress the bcr-abl oncogene and the parental clone 15v4 (in the presence of IL-3), was less sensitive. Interestingly, the effects of rapamycin could be overcome in clone 6.5 by the addition of IL-3 in the culture medium (
Fig. 1C
). This indicates that in the absence of IL-3, clone 6.5 cells are exquisitely dependent (addicted) on an intact mTOR pathway, whereas in the presence of IL-3, the IL-3/Jak/STAT signaling pathway takes over the role of the survival pathway. Indeed, addition of IL-3 increased STAT5 phosphorylation in 6.5 cells without affecting phosphorylation of S6-kinase. On the other hand, rapamycin reduced S6-kinase phosphorylation without affecting STAT5 phosphorylation (
Fig. 1B
). To further validate this system, the parental clone 15v4 was stably transfected with an shRNA against PTEN.
21
Indeed, also the resulting clone shPTEN was sensitive to rapamycin, and its effect could be abrogated by the addition of exogenous IL-3 (
Fig. 1C
). As shown also in
Figure 1C
, the milder effect of rapamycin on the non–mTOR-addicted clones Bcr-Abl and D5a could not be rescued by IL-3. Somewhat unexpected, the effects of mTOR inhibition could never be rescued by IL-3 in the TSC2 mutant D2c, probably due to the inability of this clone to phosphorylate STAT5 in the presence of IL-3.
18
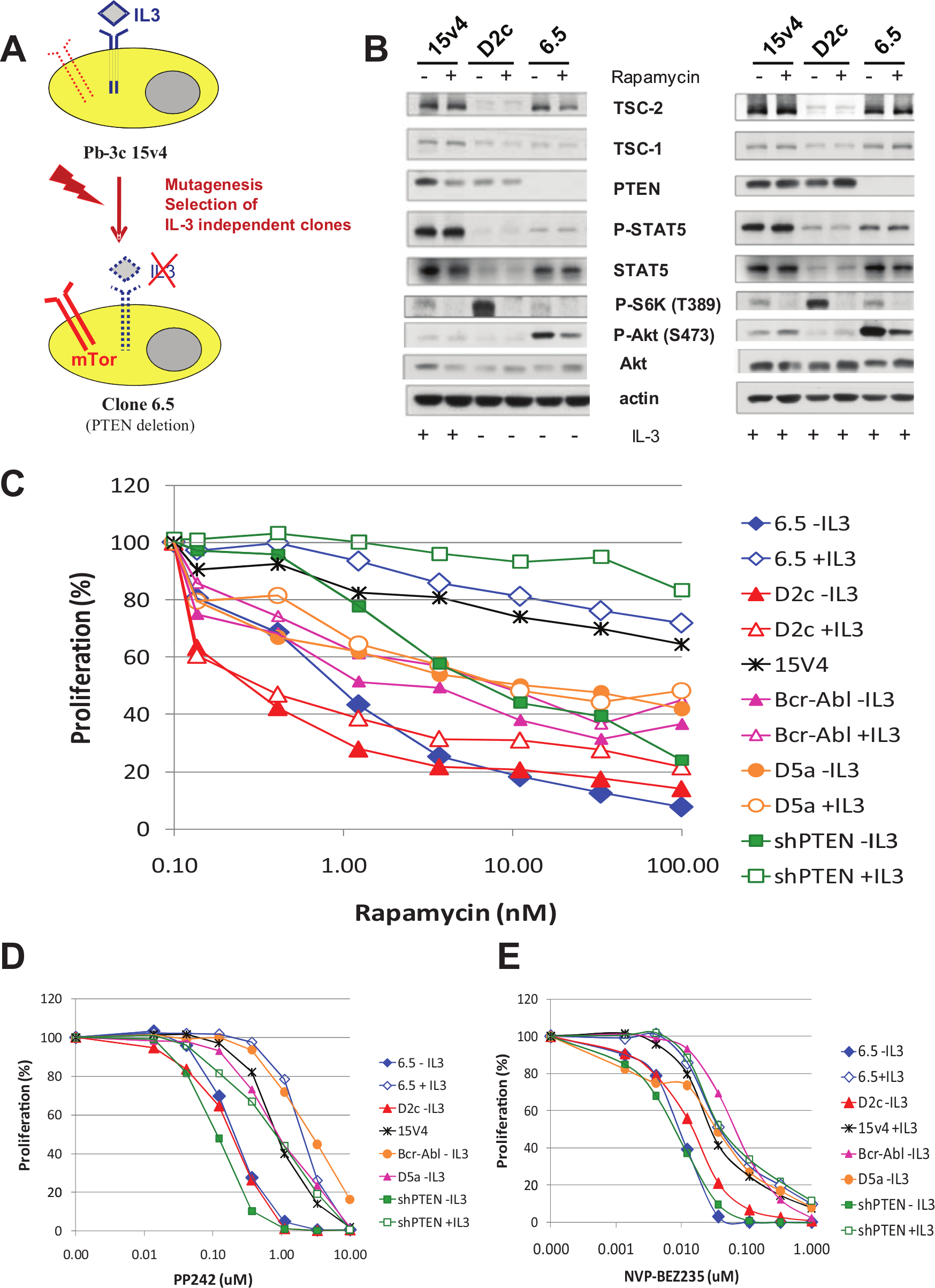
Characterization of the isogenic cell line system. (
HTS to Identify Compounds Selectively Affecting mTOR-Addicted Cells
We chose clone 6.5 for the HTS due to its high sensitivity to rapamycin and the possibility to revert rapamycin sensitivity by exogenous IL-3. A cellular growth assay in a 384-well format was established and validated for high-throughput screening. About 180,000 small molecular weight compounds were screened at 10 µM on clone 6.5 cells both in the presence and absence of IL-3. We identified 90 compounds belonging to different chemical classes, with an IC50 between 0.2 and 10 µM on 6.5 cells grown in the absence of IL-3 and a shift in IC50 on 6.5 and 15v4 cells grown with IL-3. This corresponds to a final hit rate of about 0.05%. Compounds that hit 6.5 cells irrespective of the presence of IL-3 but not 15v4 cells were not found. Details of the screen are summarized in
The 90 compounds were prioritized according to the observed IC50 shift ± IL-3, as well as to their chemical/drug-like features. Fifty-four compounds were reordered and tested on the isogenic cell line panel, with the aim to select compounds with an IC50 window ± IL-3 on 6.5 cells of at least 5-fold and that showed differential sensitivity of 6.5, D2C, and shPTEN (without IL-3) compared with D5a, Bcr-Abl, and 15v4 cell lines. Thirty-eight compounds of different chemical classes fulfilling these criteria were identified. In this report, we describe the five chemical classes with the most pronounced IC50 shift in more detail.
Hits and Their Effects on Cell Proliferation in the Isogenic Panel
Examples of hits of five compound classes are shown in Figure 2A , and the effect of these compounds on the isogenic cell line panel is shown in Figure 2B – G . Indeed, all these compounds inhibited proliferation of mTOR-addicted cell lines. IL-3 could rescue the effect of the compounds on PTEN-deficient cell lines. Subtle differences were noticed in the shapes of the dose-response curves and in the sensitivities of the different rapamycin-sensitive cell clones. D2C cells with a functional loss of TSC2 were as sensitive as 6.5 cells without IL-3 to all compounds except Cpd-4. D2C cells were much less sensitive to this compound than 6.5 cells and shPTEN cells in the absence of IL-3 ( Fig. 2E ). Cpd-1, Cpd-4, and Cpd-5 had the biggest window on 6.5 cells ± IL-3. The window between sensitive and resistant cells obtained with these compounds was bigger than the one obtained with the mTOR/PI3K inhibitor NVP-BEZ235 and similar to the one obtained with the mTOR kinase inhibitor PP242, except for Cpd2. Cpd-2 and Cpd-3 had shallow IC50 curves, similar to rapamycin, making the window between sensitive and resistant cells less clear. Cpd-5 was less potent than the other identified hits.
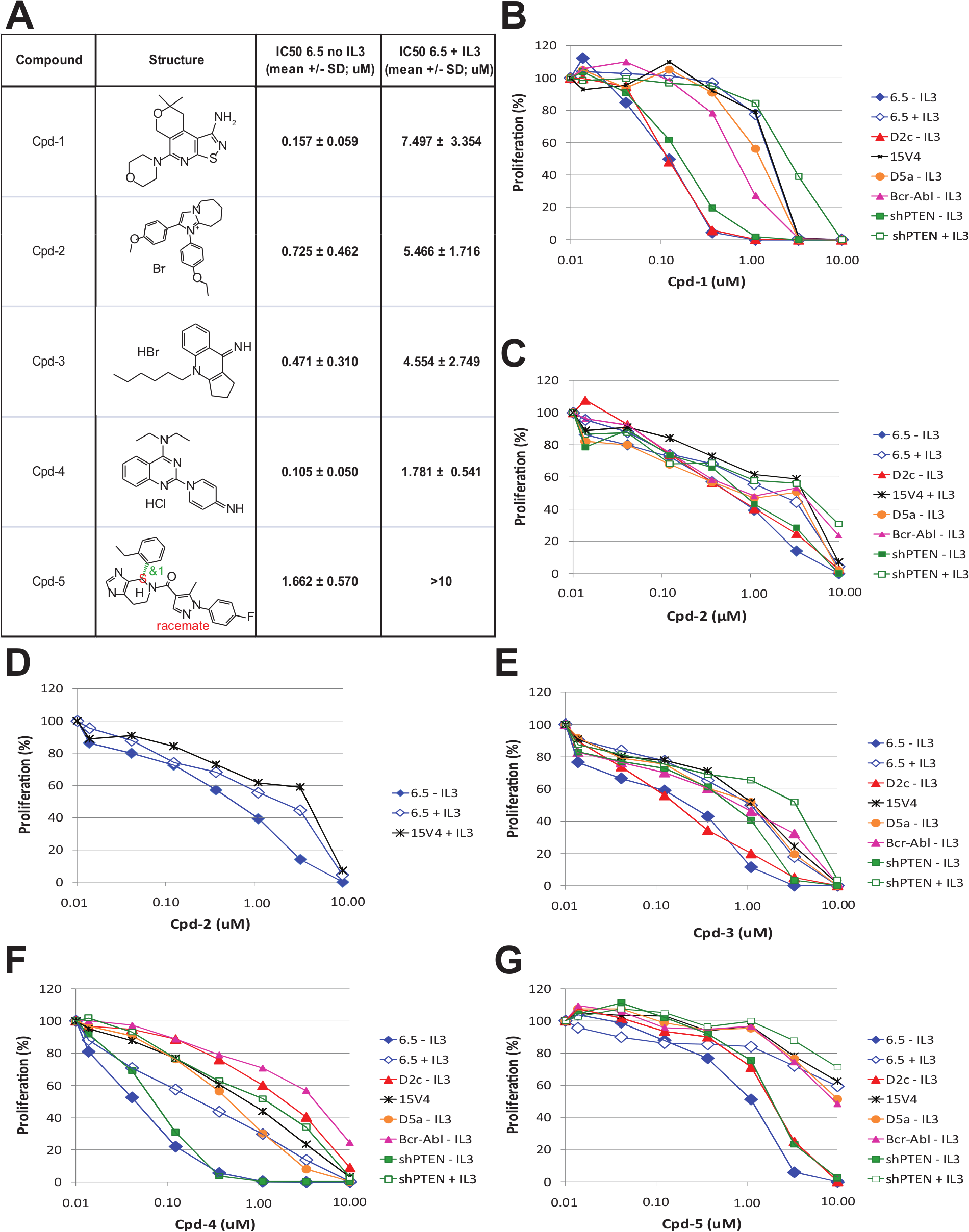
Characterization of hits in 6.5 cells. (
Mode of Action of Cpd-1 and Cpd-2
Inhibition of protein phosphorylation in 6.5 cells and kinase inhibition. In this report, we describe two hits and their mode of action in more detail: Cpd-1 (8,9-dihydro-8,8-dimethyl-5-(4-morpholinyl)-6H-Isothiazolo[5,4-b]pyrano[4,3-d] pyridin-1-amine) and Cpd-2 (1-(4-Ethoxy-phenyl)-2-(4-methoxy-phenyl)-6,7,8,9-tetrahydro-5H-midazo[1,2-a]azepin-1-ium bromide). Signaling analysis revealed that both compounds inhibited phosphorylation of S6K as well as of its downstream target, RPS6 (S6 ribosomal protein), in 6.5 cells. In addition, after a 6-h incubation, Cpd-1 also partially inhibited phosphorylation of Akt on S473, whereas Cpd-2 had no effect on Akt phosphorylation in 6.5 cells ( Fig. 3 ).
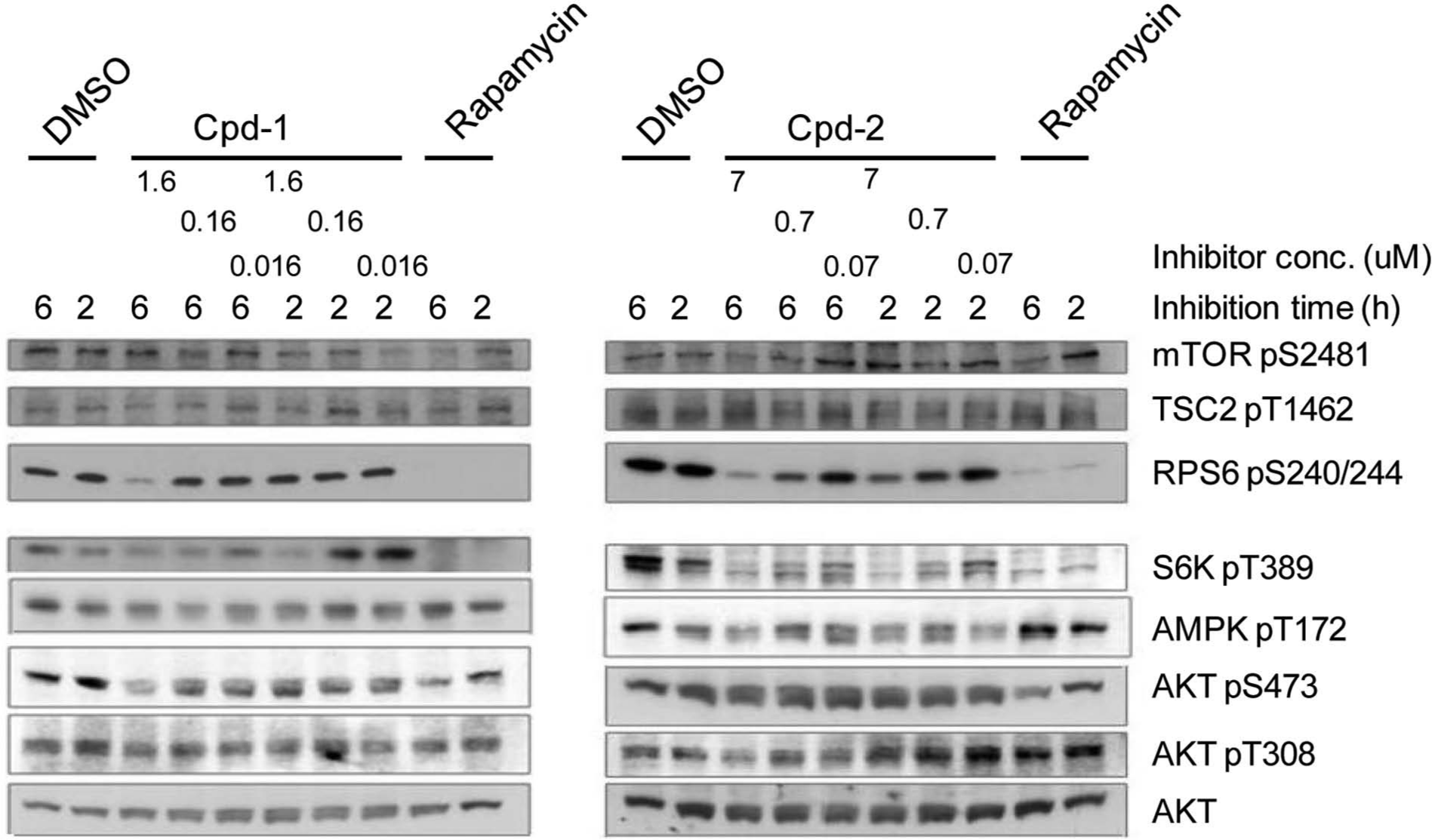
Signaling analysis of Cpd-1 and Cpd-2 in murine 6.5 cells. Signaling analysis of 6.5 cells treated with Cpd-1 or Cpd-2 for 2 or 6 h. The chosen doses corresponded to 0.1×, 1×, and 10× the IC50 of the respective compound on 6.5 cells grown without interleukin-3. Both compounds inhibited S6K phosphorylation, although Cpd-1 only at the highest concentration. Rapamycin (20 nM) was used as control, and DMSO had a final concentration of 0.1% in all samples.
Addition of IL-3 did not revert the effects of compounds 1 and 2 on the phosphorylation of mTOR pathway components (data not shown), similar to rapamycin ( Fig. 1B ).
In a next step, we asked if our compounds could be ATP-competitive kinase inhibitors. For this purpose, they were tested in a number of in vitro kinase assays related to the mTOR pathway. Interestingly, Cpd-2 had no effect up to 10 µM on all the kinases tested ( Fig. 4A ). On the contrary, Cpd-1 inhibited S6K, AMPK, and PI3K to 35% to 50% at 10 µM, with minor effects at lower concentrations. Since Cpd-2 inhibited S6K phosphorylation in cells at concentrations below 10 µM but is not an ATP-competitive inhibitor for this kinase, we explored if, like rapamycin, it requires the presence of FKBP12 to effectively inhibit cell proliferation. FKBP12 was knocked out using shRNAs in 6.5 cells. As expected, the resulting cell lines were resistant to rapamycin, even in the absence of IL-3. However, lack of FKBP12 had no impact on the efficacy of both Cpd-1 and Cpd-2 in 6.5 cells grown in the absence of IL-3 ( Fig. 4B – E ). Taken together, these data indicate that Cpd-2 is an mTOR signaling inhibitor that acts via a novel, so far not elucidated, mechanism, being neither a rapalogue nor an ATP-competitive kinase inhibitor of Akt, AMPK, S6K, mTOR, or PI3K.
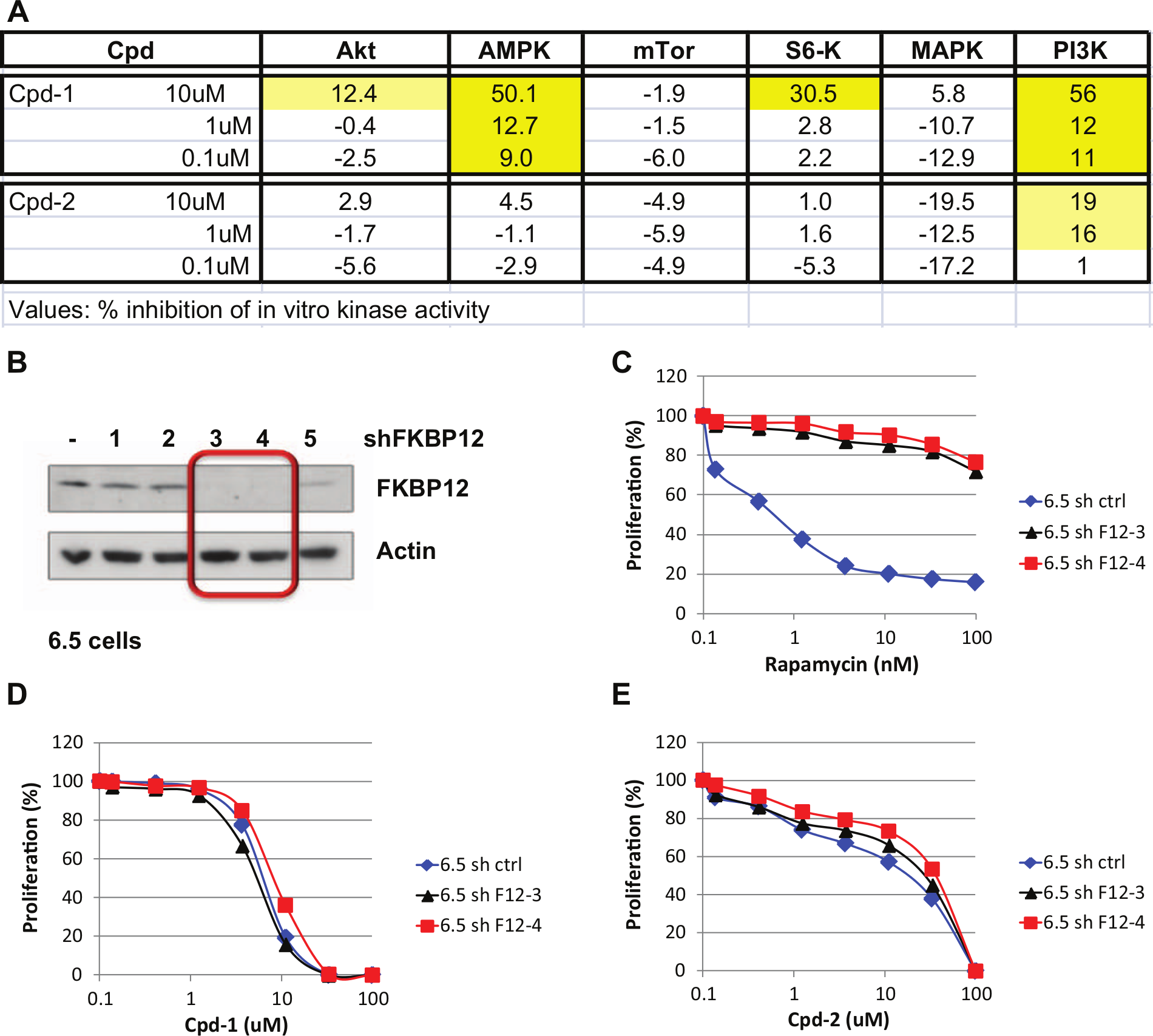
Mode of action studies of Cpd-1 and Cpd-2. (
Effect of compounds on human cells
As the isogenic cell system we used is of mouse origin, we next explored the effect of these compounds on human cells. We chose the breast cancer cell line MDA-MB-468, which has a PTEN deletion and is rapamycin sensitive.
Indeed, also in this cell line, Cpd-2 inhibits S6K phosphorylation, which translated to potent inhibition of phosphorylation of RPS6 and a reduction of cyclin D3 levels (
Fig. 5A
). We could not observe any effect of the compounds on Akt phosphorylation, probably because the cell line used has very low basal Akt phosphorylation, making it very difficult to see any inhibition. Despite some inhibitory activity in the in vitro kinase assays, Cpd-1 did not have any pronounced effect on mTOR signaling in cells, illustrated by the unchanged P-RPS6 levels (
Fig. 5A
).
Figure 5B
shows that inhibition of RPS6 as well as cyclin D3 by Cpd-2 in MDA-MB-468 cells was dose dependent, although neither phosphorylation of RPS6 nor cyclin D3 protein level was completely abrogated with 10 µM of Cpd-2. When tested in proliferation assays on MDA-MB-468 cells, Cpd-1 and Cpd-2 had an IC50 of 3.612 ± 0.493 µM and 2.593 ± 0.879 µM, respectively. Interestingly, the IC50 of compound 2 on RPS6 phosphorylation and cyclin D downregulation in MDA-MB-468 cells was around 2.5 µM (
Fig. 5B
), correlating well with the observed IC50 in proliferation assays. All five compounds were tested in proliferation assays on a number of human cancer cell lines from different cancer types and with different genotypes.
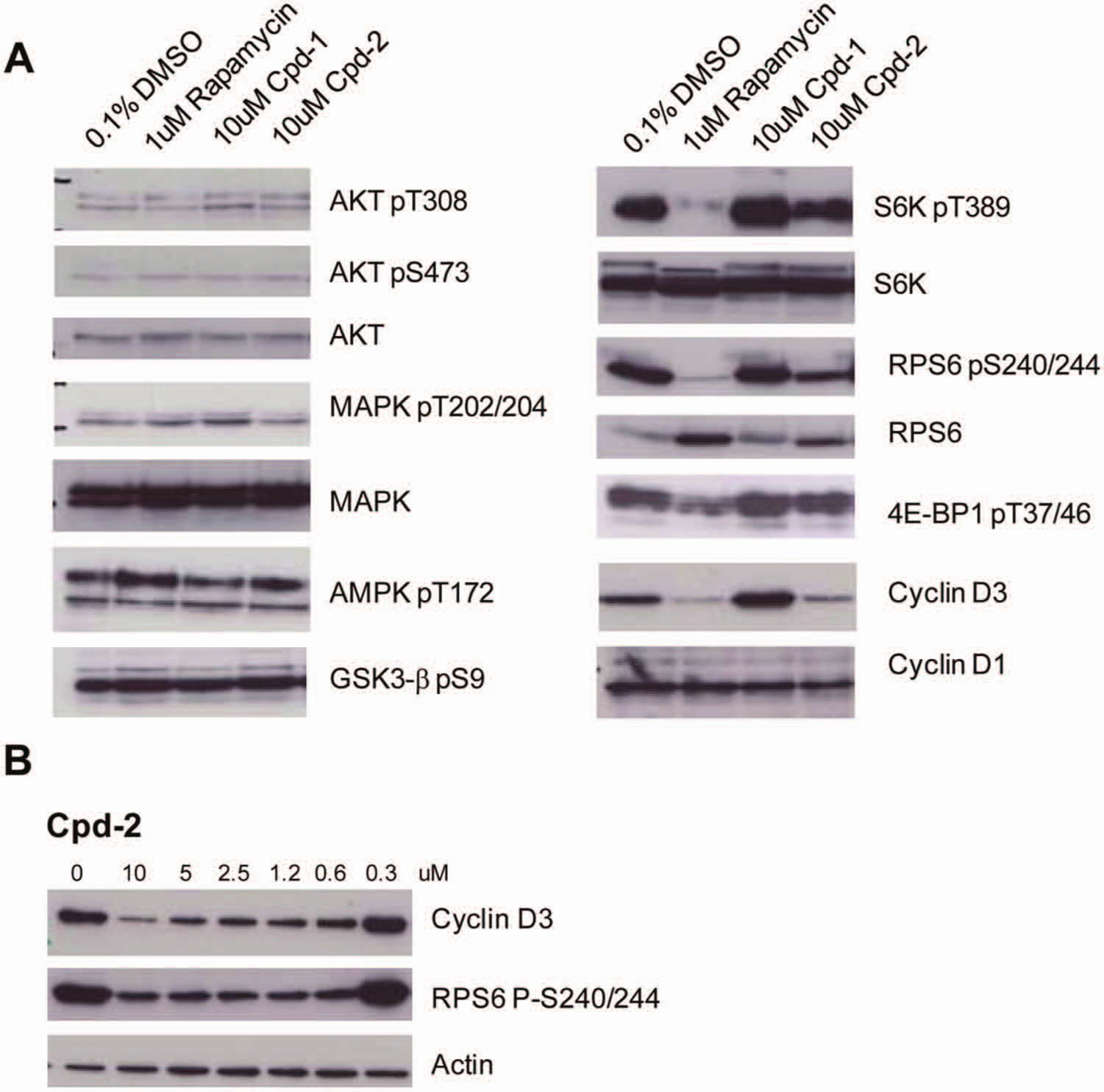
Signaling analysis of Cpd-1 and Cpd-2 in human MDA-MB-468 breast cancer cells. (
Inhibition of farnesylation and reactive oxygen species
Since one compound class was structurally similar to described farnesyltransferase inhibitors and Rheb in the mTOR pathway is farnesylated, we tested if any of our compounds would affect farnesylation. Interestingly, we found that Cpd-1 can function as a farnesylation inhibitor. As depicted in
Figure 6A
, MDA-MB-468 cells treated with Cpd-1 dose-dependently accumulated the un-prenylated form of Rap1a. This was also seen in cells treated with the farnesylation inhibitor FTI-277 but not in cells treated with Cpd-2. In addition, 6.5 cells were much more sensitive to FTI-277 in the absence of IL-3 than in the presence of IL-3 or the parental cell line 15v4 (
Fig. 6B
). The estimated IC50 on the farnesylation inhibition of 3 µM (
Fig. 6A
) correlated well with the observed IC50 in the proliferation assay (3.7 µM;
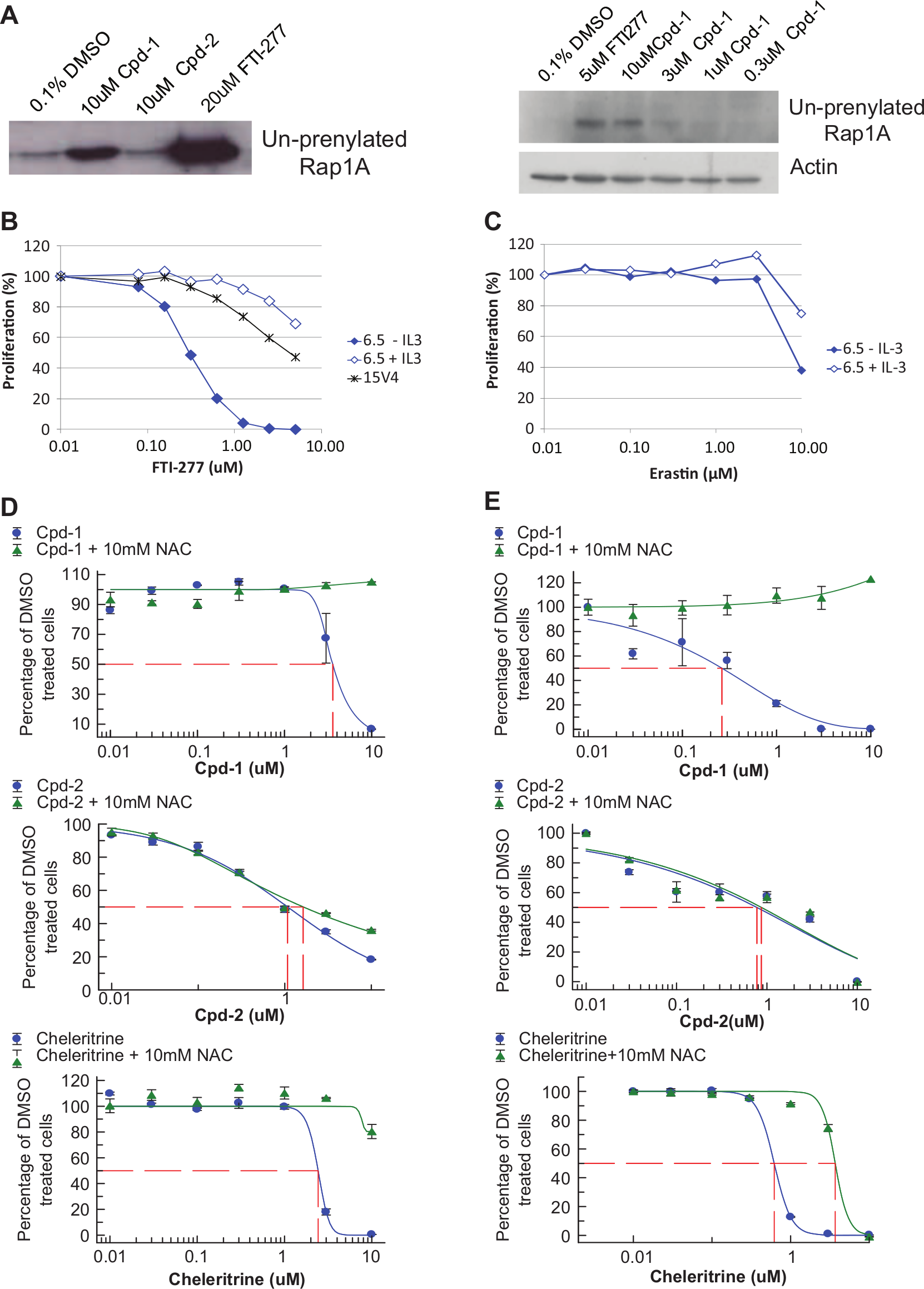
Analysis of mode of action of Cpd-1. (
It is well known that Ras also is farnesylated, that farnesylation is required for proper Ras signaling, 22 and that FTI-277 can at least partially inhibit H-Ras prenylation. 23 Thus, inhibition of Ras farnesylation could have an impact on cell proliferation. However, both cell lines, 6.5 and 15v4, carry a mutated H-Ras. If the effect of Cpd-1 and FTI-277 was mediated only by the inhibition of Ras farnesylation, we would expect equal sensitivity of 15v4 and 6.5 cells ± IL-3 to these compounds. To see whether inhibition of mutated Ras leads to differential sensitivity in 6.5 cells ± IL-3, we treated the cells with erastin, a compound described to specifically inhibit proliferation of Ras mutant cells. 17 Erastin inhibited proliferation of 6.5 cells to the same extent in the presence and absence of IL-3 (IC50 = 8.3 µM without IL-3; IC50 = 10.3 µM with IL-3; Fig. 6c ) and in a similar range to what was described by Dolma et al. 17 This suggests that Cpd-1, as well as FTI-277, in addition to affecting Ras function, also must influence other targets or pathways that lead to the observed differential sensitivity of 6.5 cells cultured with and without IL-3.
To test the additional hypothesis that our hits could inhibit cell proliferation and induce cell killing by increasing the level of reactive oxygen species (ROS) such as H2O2, we performed proliferation assays both in human MDA-MB-468 cells and in the clone 6.5 cells in the presence and absence of NAC. The antioxidant NAC has been reported to rescue H2O2-induced cell death.24,25 As depicted in Figure 6D (MDA-MB-468) and Figure 6E (6.5), indeed NAC was able to rescue inhibition of proliferation induced by the reference compound Chelerythrine 26 as well as by Cpd-1 but did not rescue the inhibition induced by Cpd-2. In summary, Cpd-1 inhibits cell proliferation of mTOR-addicted cells, at least in part by inhibiting farnesylation and increasing ROS production.
Chemical optimization
Cpd-2 is a positively charged compound. Unfortunately, removal or modulation of the positive charge resulted in fully inactive compounds, which were still charged at pH 7.4. Accordingly, fully neutral compounds with very close scaffolds were inactive. The positive charge is detrimental for oral absorption; indeed, when Cpd-2 was administered orally to rats, no absorption could be detected. Also when given intravenously, this compound had a very high clearance.
Cpd-1 is a compact, planar, and not very soluble molecule containing an isothiazole. The main efforts in chemistry were directed to improve solubility and to replace the sulfur atom. Unfortunately, attempts to render the compound more soluble resulted in loss of the difference in IC50 values ± IL-3 and replacement of the S-atom by an O- or N-atom or by enlarging the ring size with CH=CH or N=CH, leading to inactive compounds.
Mode of Action of Cpd-3, Cpd-4, and Cpd-5
Cpd-3 and Cpd-4 behaved like compounds from the Cpd-2 class and inhibited RPS6 phosphorylation in both 6.5 cells and human MDA-MB-468 cells and reduced cyclin D3 expression in MDA-MB-468 cells (data not shown). However, as described above, Cpd-4 may have a different mode of action than the others, since it does not inhibit proliferation of the D2c cell line lacking the TSC2 protein.
Interestingly, Cpd-5 inhibited RPS6 phosphorylation in 6.5 cells but did not show any effects in human cells, neither on proliferation nor on signaling. Cpd-5 is a racemic compound. We could show that only the R-enantiomer was active in 6.5 cells, but as the racemate, the R-enantiomer showed no activity in human cells (
Discussion
We have used a mouse mast cell derived isogenic cell line system to identify low molecular weight compounds that selectively affect PI3K/mTOR pathway–dependent cell clones. All the cell lines have an activated H-Ras oncogene, and different sublines, selected for IL-3–independent growth, are characterized by mutations in tumor suppressors in the PI3K-Akt-mTOR pathway, such as PTEN and TSC2. Mutations in Ras oncogenes and mutations in PTEN coexist in several human tumors. Therefore, the 6.5 cell line used for screening and compound characterization combines oncogene and suppressor gene mutations of relevance in human cancer in an otherwise “wild-type” genetic background. In addition, acquisition of growth factor/cytokine independence is a hallmark of cancer cells. The screening system has been validated with a genome-wide shRNA screen with the goal to identify genes that are essential for the IL-3–independent, transformed growth mode. Indeed, mTOR and mTORC2 were under the genes identified in the shRNA screen, confirming the specificity of the system. 21
Thus, we used the same system for a chemical HTS. We could identify compounds that fall into two main mechanistic classes. Cpd-1 is characterized by inhibition of protein prenylation, as demonstrated for the GTPase Rap1a and by production of ROS. This is in agreement with the results of the shRNA screen, where farnesyltransferase and mitochondrial genes affecting ROS balance were identified. Relevant for the Akt-mTOR pathway, the small GTPase Rheb directly regulates the activity of mTOR. Rheb is farnesylated at its C-terminus, and farnesylation is a key determinant of correct Rheb subcellular localization and assembly of a functional Rheb/mTOR complex. 27 Thus, mTOR-addicted cells, such as clone 6.5 in the absence of IL-3, should be sensitive to farnesylation inhibitors. Indeed, 6.5 cells in the absence of IL-3 were much more sensitive to FTI-277 than in the presence of IL-3 and the parental cell line 15v4.
Cells transformed with H-Ras exhibit a significant increase in ROS compared with control cells. 24 Similarly, Akt also induces the accumulation of oxygen radicals, 28 which is balanced by antioxidant systems to keep cells alive. Therefore, interfering with ROS scavengers can potentially be used to selectively kill cancer cells with high levels of Akt activity, such as PTEN null cells. Cpd-1 might exactly do this and lead to abnormal increases in ROS, which selectively kill 6.5 cells. In 6.5 cells, Cpd-1 also reduced mTORC2-mediated S473 phosphorylation of Akt, similar to long-term treatment with rapamycin, which could further contribute to an imbalance of antiapoptotic mechanisms. Inhibition of S473 phosphorylation is unlikely to be due to direct inhibition of the mTOR catalytic site, since none of the compounds described here has been found to inhibit mTOR catalytic activity.
The mechanism of three other compound classes is characterized by inhibition of phosphorylation downstream of mTORC1, such as S6K/RPS6 and, to some extent, 4EBP1, as exemplified by Cpd-2. This inhibition is most likely not due to direct inhibition of the catalytic sites of kinases, nor does it require the rapamycin binding partner FKBP12. Therefore, the compounds identified in this screen, even though closely overlapping mechanistically with rapamycin, appear to work through a novel mode of action. For example, in contrast to rapamycin, Cpd-2 reduced phosphorylation of Akt at T308 with longer exposure times. Similar to rapamycin, Cpd-2 potently downregulated cyclin D3 in human MDA-MB-468 cells. In 6.5 cells, both rapamycin and Cpd-2 had only minor effects on cyclin D3 expression (data not shown). Downregulation of cyclin D3 leading to cell cycle arrest by rapamycin and NVP-BEZ235 has been shown in different cell types.29,30 Indeed, PTEN appears to be a general negative regulator of cyclin D expression, 31 and D cyclins are overexpressed in many cancers. 32
All compounds with the exception of Cpd-4 consistently inhibited proliferation of cells with altered or downregulated proteins in the PTEN-Akt-TSC-mTOR pathway. This inhibition could be overcome by treating cells with IL-3 in all lines but not in those lacking TSC2, similar to what was observed with other mTOR inhibitors. The specificity of the compounds for PTEN-mTOR–addicted cells is shown by the reduced sensitivity of Bcr-Abl–transformed mast cells as well as the STAT-5–dependent clone D5a.
Although Cpd-4 had the same effect as Cpd-2 and Cpd-3 on signaling, interestingly, this compound did not inhibit proliferation of D2c cells lacking the TSC2 protein, indicating that its main inhibitory effect must be upstream of TSC2.
Cpd-5 was the least potent compound on mouse 6.5 cells but showed a big (10-fold) window to 6.5 cells in the presence of IL-3. Unfortunately, this compound, even at high doses up to 30 µM, did not affect either PI3K-Akt-mTOR signaling or proliferation in human cells. Antiproliferative activity was also observed in a rat cell line but was not followed up (data not shown). It may thus be that this compound affects a rodent-specific target and/or that much higher doses than the one tested have to be used to see an effect in human cells. It has to be taken into account that with all compounds identified here, we observed a shift in potency from the mouse isogenic cell line system to human cancer cell lines. A possible explanation is that the murine system, harboring only two known genetic alterations (PTEN and H-Ras), is more dependent on an intact PI3K/mTOR pathway than the genetically diverse human cancer cell lines. Indeed, when cultured in the absence of IL-3, 6.5 cells undergo apoptosis in the presence of nanomolar amounts of rapamycin. 21 In contrast, it is impossible to induce apoptosis with rapamycin in human cancer cell lines, and micromolar concentrations of rapamycin are required to see (partial) growth inhibition. We therefore assume that despite PTEN silencing and Ras activation, additional mutations in human cancer cells might overrun components of the Akt-mTOR pathway and therefore render the cells less addicted. For instance, the human colon cancer line HCT-15, which is characterized by PI3K, Ras mutations, and APC mutations, is completely resistant to the compounds described here but still highly sensitive to the mTOR inhibitor PP242. APC mutations have been described to exert dominant effects on cell proliferation and checkpoint control 33 and might therefore be able to overrun the Akt-mTOR-pathway selective inhibition by the compounds described in this study but not of the mTOR kinase inhibitor PP242. On the other hand, it has been recently described in breast cancer cells that cells with mutations in the PI3K gene are highly sensitive to mTOR inhibitors, while cells with PTEN mutations are not, suggesting that the functional consequences of these two mechanisms of activation of the mTOR pathway are quite distinct. 34 The compounds described here may just have the reverse specificity and preferentially affect cell lines with PTEN mutations. The fact that these compounds are active only on a subset of human cancer cell lines indicates that they act in a context-specific manner. More experiments involving larger cell line panels will be needed to identify the most sensitive genotypes as well as potentially synergistic drug combinations. Nevertheless, it may be difficult to correlate sensitivity to a defined genotype under standard tissue culture conditions. Using our limited panel of cell lines, it was also not possible to identify the genotype with the highest sensitivity to the mTOR kinase inhibitor PP242. Interestingly, this inhibitor showed the highest activity on RPMI-8266 and A498 cells, which do not harbor any mutations in the mTOR/PI3K pathway.
Recently, another phenotypic screen was published that identified one compound preferentially inhibiting proliferation of cells with PTEN deletions. 35 In this screen, the authors used somatic gene targeting of the human colorectal cancer cell line HCT-116 to generate isogenic PTEN−/− and PTEN+/+ clones. The advantage of this system, compared with ours, is that it is of human origin. However, we believe that because HCT-116 cells harbor mutations in several genes on top of the PTEN and Ras genes and display 3% to 7% polyploidy, they may not be completely dependent on an intact PI3K/mTOR pathway. Thus, it may be more difficult to find compounds that specifically inhibit proliferation of mTOR-pathway addicted cells.
In general, the use of isogenic cells for phenotypic compound screens bears several challenges: the compounds need to be cell permeable, the establishment of a structure-activity relationship is difficult without a defined molecular target, and the resulting hits may be very context dependent. On the other hand, generally toxic compounds, affecting also the control cell line, are eliminated very early, and there is the possibility to identify compounds affecting more than one target (poly-pharmacology). To minimize the challenges mentioned above, we propose to choose a screening system that recapitulates aspects of the patient’s disease as closely as possible, as well as to select compounds that already have drug-like properties and are easily amenable to optimization.
Taken together, our results show that is possible to use phenotypic screening of isogenic cell lines to identify compounds targeting cells with a defined genotype (comprising also loss-of-function mutations). The identified compounds have a novel mode of action and target PI3K/mTOR-pathway addicted cells via different mechanisms affecting either the mTOR pathway directly or other accessory but necessary pathways. These findings may be translated to specific combinations of drugs interfering with the individual targets or pathways affected by the compounds described in this work.
Footnotes
Acknowledgements
We thank Stefanie Keller (BioFocus, Allschwil Switzerland) for performing the HTS, Charles Betz for identifying the frame shift mutation leading to the absence of functional TSC2 protein in the D2c cells, and Tabitha Bucher for help with proliferation assays.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: LWP, AK, MG, PG, and UR are employees of Actelion Pharmaceuticals Ltd.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by a grant from the Swiss Commission for Technology and Innovation (CTI) [grant number #8963] to CM, MNH, and UR.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.