
Abstract
Dysfunctions at the level of RNA processing have recently been shown to play a fundamental role in the pathogenesis of many neurodegenerative diseases. Several proteins responsible for these dysfunctions (TDP-43, FUS/TLS, and hnRNP A/Bs) belong to the nuclear class of heterogeneous ribonucleoproteins (hnRNPs) that predominantly function as general regulators of both coding and noncoding RNA metabolism. The discovery of the importance of these factors in mediating neuronal death has represented a major paradigmatic shift in our understanding of neurodegenerative processes. As a result, these discoveries have also opened the way toward novel biomolecular screening approaches in our search for therapeutic options. One of the major hurdles in this search is represented by the correct identification of the most promising targets to be prioritized. These may include aberrant aggregation processes, protein-protein interactions, RNA-protein interactions, or specific cellular pathways altered by disease. In this review, we discuss these four major options together with their various advantages and drawbacks.
Introduction
In eukaryotes, proper regulation of gene expression requires the coupling of many processes that include transcription, pre–messenger RNA (mRNA) splicing, mRNA editing, surveillance, stability/transport, and finally translation. 1 All these processes were initially considered functionally separate units, with each process regulated by a distinct set of factors. However, research in the past two decades has uncovered a very high degree of coupling, and it is now commonly accepted that nuclear events occurring very early in nascent RNAs can condition their ultimate fate in the cytoplasm.2–4 Most important, this conditioning is not unidirectional, and we are also just starting to understand the many ways in which gene expression can be modulated by dynamic changes in chromatin structure driven by small and noncoding RNAs.5–7 The practical consequence of this extremely complex situation is that RNA processing represents a crucial regulatory node in the control of gene expression. This is especially true in higher organisms and in those organs that display the highest complexity in terms of developmental plasticity, protein isoform production, and noncoding RNA expression.
In keeping with this view, RNA processing events have been shown to play a major role in the regulation of brain development and normal functioning.8–11 This higher complexity also means that defects at the level of RNA metabolism can often represent a major cause of neuronal and synaptic impairment, eventually leading to disease. Indeed, a connection between RNA metabolism and neurodegeneration has always been known to be important for monogenic diseases that involve the nerve system, such as neurofibromatosis. 12 At a more general level, high-throughput analyses have recently shown that alterations in general RNA processing events can be observed in all the most common forms of neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration (FTLD), Parkinson disease (PD), and Alzheimer disease (AD).13–16
Widespread alterations in RNA metabolic pathways usually include several overlapping effects. First, there is a physiological decrease in RNA processing efficiency that comes with the normal aging process. 17 In disease-affected individuals, on top of these natural changes, there are also disease-specific losses in RNA processing efficiency. It is these specific losses that are currently thought to considerably accelerate and worsen the natural decline.
The possible causes for this RNA-related damage can occur in many different ways, such as the formation of aberrant RNA foci, bidirectional transcription, or the production of toxic RNAs and their resulting proteins. 18 In many cases, these losses also originate from impairments of macromolecular complexes important for RNA processing, such as the spliceosome 19 or the microRNA processing systems. 20
In most of these cases, functional alterations of these machineries can derive from changes in the primary sequence, intracellular localization, or aggregation status of various RNA binding proteins (RBPs) that participate in the assembly and activity of these complexes. The identification of these RBPs and the way they affect RNA processing functions have therefore become a major target of recent research in neurodegeneration. 21 Once identified, the RBPs responsible for the dysfunctions have immediately become the preferential targets of biomolecular screening analyses aimed at evaluating their possible use as new biomarkers, to follow disease onset/progression, or as promising therapeutic targets to slow/reverse disease.
RBP Proteins in Neurodegeneration
RBP proteins play an essential role in providing the connection between all the gene expression steps that lead to the production of a mature RNA. 22 Most important, RBPs tend to assemble in distinct multimeric complexes that control different steps of specific RNA life cycles.23,24 In this way, by varying their specific RBP composition, the various macromolecular systems form a bridge among the different steps and, at the same time, display different functional properties.
In terms of numbers and quantity, the RBPs responsible for creating the “backbone” of these multimeric complexes belong for the most part to the heterogeneous ribonucleoprotein (hnRNP) family. 25 From a structural point of view, all hnRNPs contain one or more RNA binding domains that allow specific binding to RNA sequences. As determined in many studies, they also contain specific sequences of variable composition, such as arginine-glycine-glycine (RGG) boxes that mediate their biological functions.26,27 This fundamental role played by hnRNPs is especially highlighted in neurons, where there is an extremely high level of differential RNA expression that is mainly generated through the cooperation of these ubiquitous hnRNP proteins with other tissue-specific RNA binding factors.28–30
At present, the most studied hnRNP proteins that have been shown to play a major and direct role in neurodegenerative processes are represented by TDP-43,31,32 FUS/TLS,33,34 FET proteins, 35 and very recently two hnRNP A/B family members. 36 These proteins, it is important to note, do not act alone in mediating neurodegeneration. For example, the recent description of the presence of hexanucleotide expansions in the C9orf72 gene associated with disease37,38 has also raised the possibility that the effect of pathological RNA foci may contribute to sequester additional RBPs and thus contribute to influence the onset and progression of disease. At the moment, very little is known about the interplay between all these various factors and the way they may affect disease. Nonetheless, these findings have sparked extensive research in the connection between RBPs and neurodegeneration. Indeed, they are likely to become a major theme in future research on this topic, as discussed in several recent reviews.39,40
Most important, these findings have triggered novel screening, diagnostic, and especially therapeutic possibilities, schematically summarized in Figure 1 , and have already provided a whole new spectrum of targets that are expected to improve our fight against ALS, frontotemporal dementia, Alzheimer disease, and other neurodegenerative diseases. From the moment of their discovery, in fact, knowing which RBPs are involved in a particular neurodegenerative disease has allowed population screening studies to look for mutation carriers and establish genotype-phenotype correlations41,42 ( Fig. 1 ). In parallel, their eventual aberrant presence in easily available patient samples such as plasma, peripheral blood mononuclear cells (PBMCs), or cerebrospinal fluid (CSF) has also been used to try and set up better diagnostic criteria43–45 ( Fig. 1 ). Most important, knowing the identity of the responsible RBPs has allowed the development of model systems with the aim of mimicking the pathological features observed in affected individuals.46–50 The development of these models, together with a better knowledge of the biological functions of proteins involved, are key factors that pave the way to hypothesize novel therapeutic options ( Fig. 1 ).
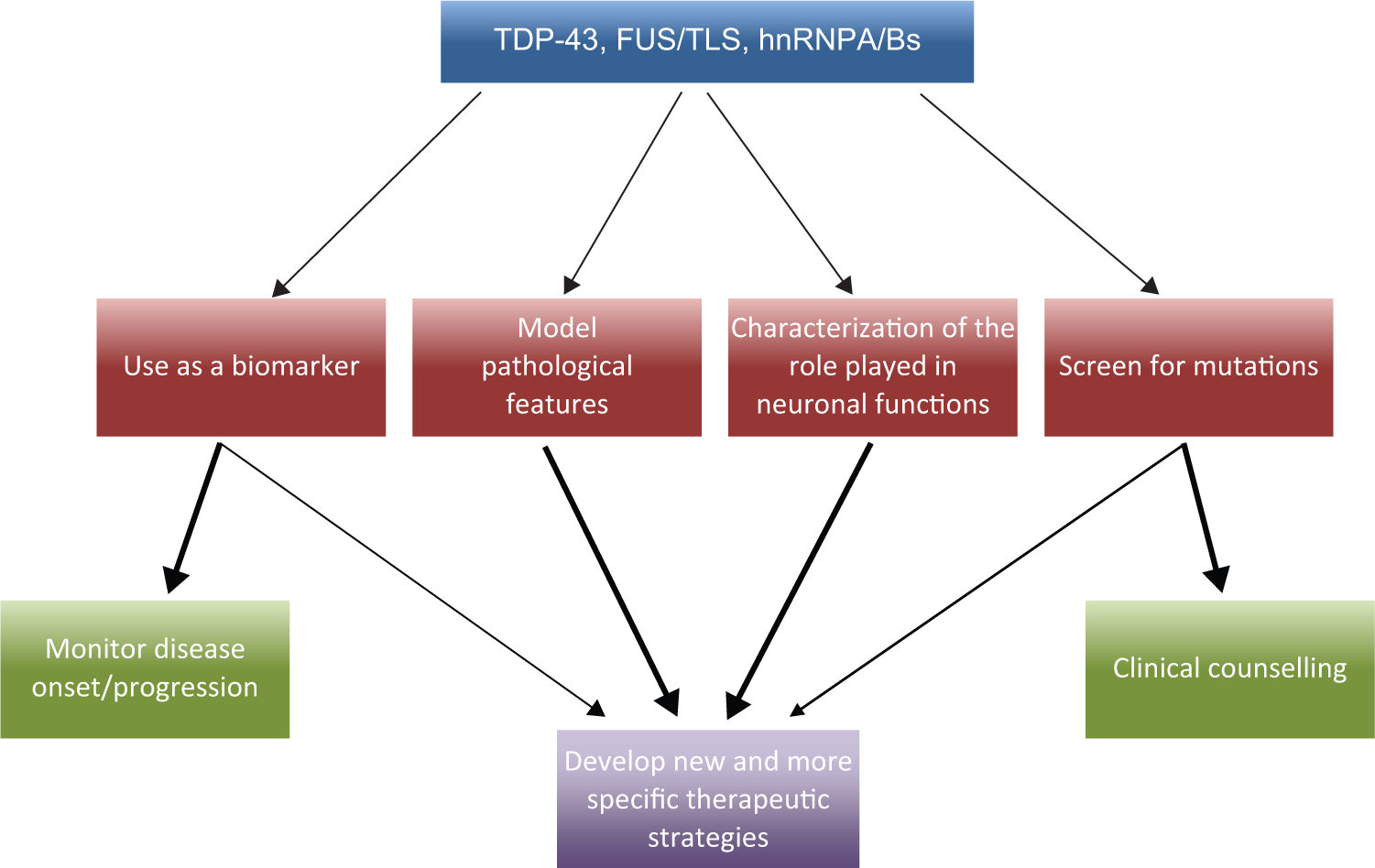
Targeting RNA binding proteins in neurodegenerative diseases. This figure shows the possible pathways through which the study of RNA binding proteins such as TDP-43, FUS/TLS, or the heterogeneous ribonucleoprotein (hnRNP) A/B factors involved in neurodegeneration can provide novel diagnostic, screening, and therapeutic approaches to researchers. The various arrows indicate what are the preferential study areas that should be targeted and which aspects of our knowledge will benefit the most from these kinds of studies.
Finally, the characterization of the RNA and protein interactors of different hnRNPs might be helpful for defining the extent of overlaps among the regulated pathways. In this context, for example, recent studies have found that a subset of transcripts harboring several intronic binding sites for both TDP-43 and FUS/TLS contain exceptionally long introns and encode for proteins crucial for normal neuronal function. 51
There are four major areas where efforts could be directed in future screening studies (schematically summarized in Fig. 2 ). First, the foremost major feature of these proteins to be targeted might be the pathological features displayed by these RBP proteins in disease (i.e., aggregation, aberrant cytoplasmic localization, etc.). Another potential therapeutic approach might consist of targeting specific protein-protein and RNA-protein interactions that may become altered in the disease state ( Fig. 2 ). Finally, rather than focusing on single events (that may be difficult to pinpoint accurately at least in this early stage of research, as will be further discussed), one could also hypothesize to target the major metabolic pathways that become impaired following the aberrant localization/expression of the various RBPs ( Fig. 2 ).
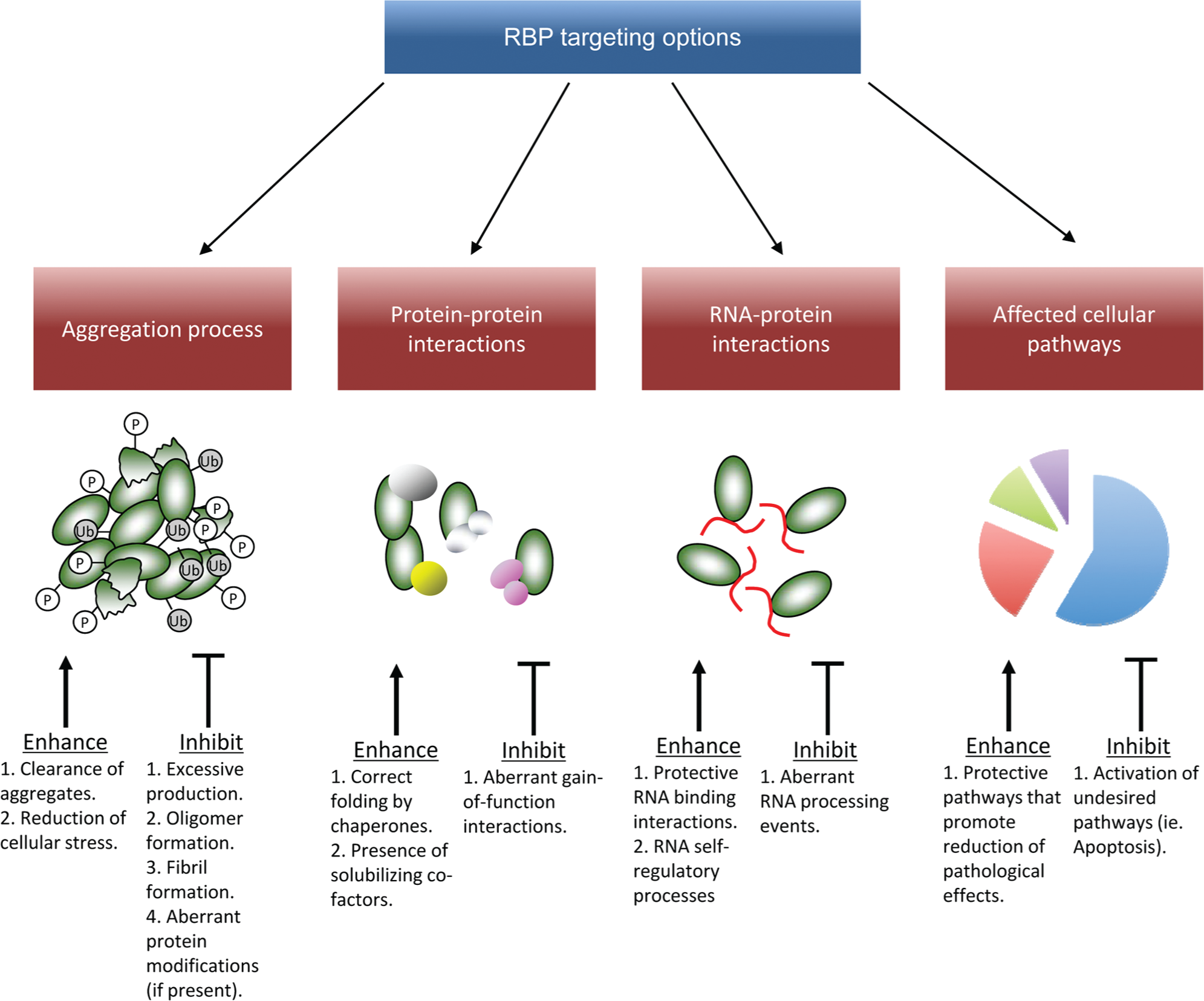
Possible strategies to target RNA binding proteins (RBPs) involved in neurodegeneration. This figure provides a schematic overview of the various options that can be used to target various aspects of RBP properties. These range from targeting the aggregates formed by these proteins to affecting their protein binding, RNA binding, or autoregulatory properties.
Since TDP-43 is one of the oldest known RBPs directly involved in neurodegeneration, there is a wealth of information regarding its major biological properties. Much is now known regarding its global RNA binding/protein binding properties and the pathways that can become predominantly affected following its aberrant aggregation in the cytoplasm of neuronal cells.45,52 Therefore, in this work, particular attention will be devoted to this protein when discussing the various approaches described above. Nonetheless, because of their biological and structural similarities, most of the considerations that will be made regarding TDP-43 will generally apply to all RBP proteins of similar composition/activity.
Targeting Aberrant RBP Aggregation
Aggregation is one of the most distinguishing features of many neurodegenerative diseases. As a result, aggregate formation should represent one of the most obvious and preferred targets for the development of novel therapeutic effectors. Unfortunately, the events that regulate this process are still hard to define, in terms of their biological significance and of the factors/pathways that trigger this process. 53
Aggregation involves several successive steps ranging from protein misfolding, oligomerization, and finally the eventual formation of ordered or amorphous fibrils/filaments (for a detailed review on the subject, the reader is referred to Bartolini and Andrisano 54 ). Based on these experimental findings, and depending on the aggregated protein under study, many strategies have been developed to try and stop this process. These strategies include all the various steps of aggregate formation: from approaches aimed at inhibiting basic protein production levels, inhibiting all the various intermediate steps that lead to fibril or filament formation, or achieving the resolubilization of already formed, mature aggregates. 54
To develop a successful therapeutic strategy against any aggregation process, therefore, it is important to obtain information regarding the composition of the aggregates, the genetic or environmental factors that mediate their formation, and especially their significance with regard to the pathological process. As will be described below, this is not an easy or straightforward process even for well-studied proteins such as TDP-43.
The most common sign of ALS/FTLD pathology is the presence in the patient’s neurons of nuclear and cytoplasmic TDP-43 aggregates that are ubiquitinated, hyperphosphorylated, and cleaved to generate C-terminal fragments (CTFs). 55 The presence of the cytoplasmic aggregates is often accompanied by nuclear clearance of the protein, an observation that may help to explain the RNA processing defects observed in aggregate-bearing cells (see below for more discussion on this subject).
At present, the direct pathological connection of the aggregates with human disease is still unclear.56,57 Indeed, aggregates may be considered to cause a “loss-of-function” effect by sequestering active TDP-43, could be inherently toxic (“gain of function”), or could even represent a neutral marker of disease (although based on recent results from cellular and animal models, this third possibility is rather unlikely, at least for TDP-43 and FUS/TLS). Regarding the pathology, it should also be considered that loss- and gain-of-function effects are not mutually exclusive and may both contribute to disease, as recently reviewed.58,59 Strictly speaking, in case of TDP-43 disease-associated mutations, the presence of aggregates may not even be required to result in neurodegeneration.60,61 Finally, another very important function of the TDP-43 aggregates in disease could be their ability to form “cross-seeding” units that allow the disease to spread from cell to cell and neighboring brain regions in a prion-like manner, 62 as hypothesized for many other RBP proteins. 63
Obviously, providing an answer to these questions may have huge consequences on future therapeutic approaches if we consider that the presence of TDP-43 aggregates remains to this date the most distinguishing feature of the human pathology.
At present, the major problem of investigations in this direction is represented by our inability to mimic human aggregates in a satisfactory manner. In cell cultures, TDP-43 aggregates have been successfully obtained using TDP-43 variants that lack particular functional domains such as NLS signals, the C-terminal tail, or by directly transfecting C-terminal fragments.64–67 The major drawback of these approaches is that results cannot be easily compared with each other as the various studies often use different cell lines and transfection conditions (in addition, most studies rely on transient transfections whose differing efficiency may also lead to variable results). Researchers in this field are very much aware of these limitations, and as a consequence, a lot of effort has gone to establish a model system of TDP-43 pathology in various animal models, especially rodents.46,68,69 Unfortunately, most models obtained so far do not display TDP-43 aggregates,46,70 and even those that do71,72 generally fail to satisfactorily mimic human pathology.73,74
More recently, several unconventional approaches have also been attempted. For example, it has been shown that in cell lines and primary rat neuronal cultures, the introduction of tandem repeats carrying the 331-369 Q/N region of TDP-43 coupled to a reporter protein (enhanced green fluorescent protein [EGFP]) can trigger the formation of aggregates that can sequester endogenous TDP-43 in the cytoplasm and display most of the characteristics of human aggregates. 75 Furthermore, formation of these aggregates can also be enhanced by pathological missense mutations that interfere with hnRNP A/B binding to this Q/N-rich region. 76 The advantage of such systems, beside their ability to trigger TDP-43 aggregation, is that they achieve this aim without affecting basal TDP-43 expression levels.
Following the development of a satisfactory model of aggregation, the successive step is then to find/develop compounds that are capable of affecting its formation/solubilization. At present, this is still very much a process of trial and error, and several compounds have already been tested for an effect on TDP-43 aggregation, as recently reviewed by us. 45 At the moment, the major drawback in screening for effective molecules capable of slowing down or solubilizing TDP-43 aggregates comes from the fact that compounds that work well in a particular model may fail to have any protective effect in a different model, thus making their validation more difficult for more advanced pharmaceutical studies. Just to provide an example, methylene blue has been described to be effective in a transient cellular model of TDP-43 aggregation 77 and in two Danio rerio and Caenorhabditis elegans models of TDP-43 toxicity 78 but had no protective effect in a mouse transgenic model expressing a TDP-43 carrying the disease-associated G348C mutation. 79 The reasons for these discrepancies are currently unknown. One possibility is that insufficient exposures were achieved with the compounds used or that in simpler organisms, life span and neuroprotection may be uncoupled from each other.
Taken together, these observations suggest that RBP aggregate model systems should always be thoroughly validated to make sure that they accurately reflect, as much as possible, the human situation. Only when this aim will be achieved, the use of biomolecular screening approaches will stand the best chance for identifying compounds worthy to be carried forward toward a more advanced clinical setting. Second, it should also be very important to deeply characterize the molecular regions of the RBP protein of interest that are involved in the aggregation process. This will allow the “intelligent” design of small molecules capable of interfering with this process in a highly specific manner, hopefully leaving unaffected the protein’s normal functions.
Targeting Protein-Protein Interactions
The modulation of protein-protein interactions (PPIs) has recently become a reality in the biomolecular screening field. 80 In particular, several strategies have been recently developed to improve our ability to modulate these interactions by using a variety of compounds commonly referred to as PPI modulators (PPIMs).81,82 At the practical level, modulation by PPIMs can be achieved using two major strategies. The first option is to create a steric hindrance at the regions of the proteins involved in the reciprocal interactions (and in this case, they are called orthosteric PPIMs). The second option, on the other hand, is for the PPIM to bind away from the surface directly responsible for the interaction but in a critical position to induce/prevent conformational changes required for the PPI interaction to take place. In this case, the PPIMs are referred to as allosteric PPIMs.
As previously described, many hnRNP proteins have the ability to connect with each other and with other factors to form multifunctional complexes. The best-characterized binding partners of TDP-43 are several members of the hnRNP A/B protein family that have been shown to be very important for the splicing inhibitory properties of TDP-4327,83 (see also below). In addition to hnRNP proteins, many others have been proposed to be possible, and proteomic studies performed principally in culture cell lines such as HeLa 84 and HEK-293T 85 have shown that TDP-43 potentially displays several hundred potential interactors that could well explain its different functional properties within the cell. In addition to these very focused studies, there is also already a huge wealth of information coming from general studies aimed at elucidating the interactome profile of human proteins.
Therefore, by focusing on direct (physical) and indirect (functional) associations derived from published high- and low-throughput screening analyses in different databases,86–88 many TDP-43 interactors at the protein level can be outlined ( Table 1 ). The studies of direct associations are of great interest because in several cases, they have allowed the identification of the domains crucial for the interaction with TDP-43. In this context, the HRDC (spanning the residues 503–583) domain of human exosome component 10, 73 the exonuclease domain (encompassing residues 1–280) of Xrn2/Rat1, 73 the UBQLN UBA domain, 89 the POZ/BTB domain of NAC1, 90 and the C-terminal tail of hnRNP A2 spanning residues 288 to 341 75 all represent some paradigmatic example.
TDP-43 Protein-Protein Interactions.

The methods for physical interactions are classified according to the BioGRID experimental evidence codes (http://wiki.thebiogrid.org/doku.php/experimental_systems).
MS, mass spectrometry.
Finally, when evaluating a protein interaction profile, it is also important to consider ad hoc studies. For example, different mutants of VCP (R95G, R155H, R155C, R191Q, and A232E) have been found to be colocalized with TDP-43 in the nucleus, leading to a more granular distribution of TDP-43, an observation that suggests an interplay between the two proteins. 91
The comparison of two proteomic studies specific for TDP-43 with the other high- or low-throughput proteomic studies reported in Table 2 highlights the interaction of TDP-43 with hnRNPs (in particular, A1, K, and R) and with FUS/TLS ( Fig. 3A ). These observations not only strengthen the hypothesis that these associations are important for the proposed TDP-43 functions in RNA metabolism but also suggest that alterations of any of the partners recruited in these interactions can perturb common molecular pathways in which these proteins are involved, leading to neurodegenerative disease.
Cellular Pathways Affected by TDP-43 Protein-Protein Interactions. a

Functional enrichment analysis was performed using DAVID Bioinformatics Resources (Frederick, MD).
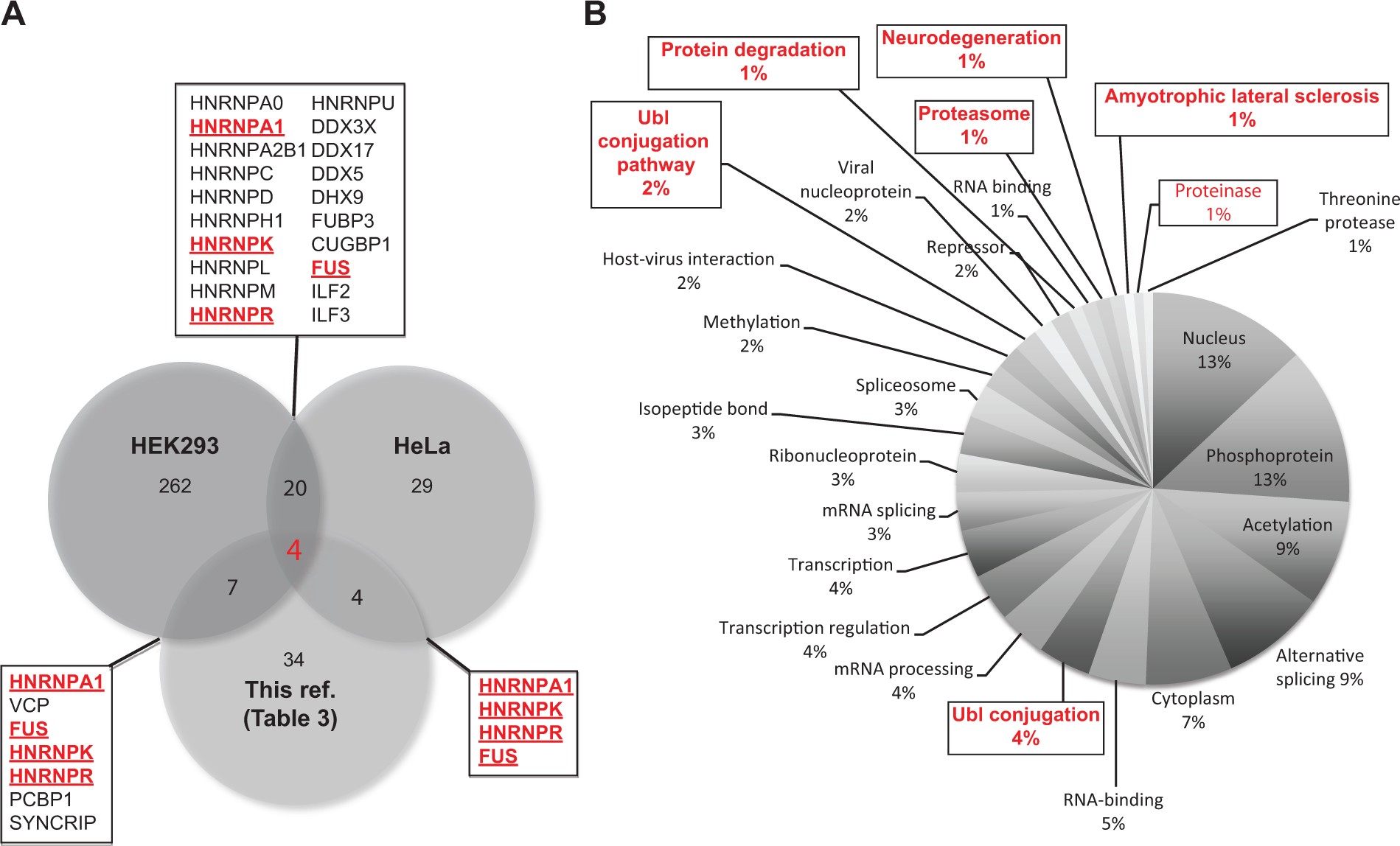
Overlapping comparison of various TDP-43 protein-protein interactions shown as a Venn diagram and schematic diagram of the cellular pathways affected by TDP-43 protein-protein interactions. The data sets analyzed in
It is also to be expected that many of these interactions will probably be protective against aberrant aggregation processes by helping to maintain the protein’s localization and solubility. Of course, the challenge currently lies in identifying and characterizing these protective interactions. Indeed, recent evidence in this direction has been obtained with regard to the interaction of TDP-43 with hnRNP A/Bs through its aggregation-prone Q/N-rich region.75,76,92 It is therefore expected that effectors capable of stabilizing this interaction might protect against the aberrant processing of this protein ( Fig. 2 ), and further studies will be aimed in this direction. In a specular but opposite manner, it has also been reported that the affinity for FUS/TLS of a mutated TDP-43 is much higher than for the wild-type protein, 84 suggesting that this interplay may be important for disease. Hence, small molecules capable of interfering with this aberrant interaction might prove beneficial to reduce mutated TDP-43 pathology.
Finally, a growing body of evidence also suggests that several of the TDP-43 interactors so far validated can also be independently implicated in the pathogenesis of similar disorders since a recent report has found that mutations in hnRNPA2/B1 and hnRNPA1 can also lead to multisystem proteinopathy and ALS. 36 Therefore, the hypothesis that an alteration in the protein-protein interaction profile of a single protein might affect eventual disease-associated functions of its partners is far more than a simple speculation.
Targeting RNA-Protein Interactions
Several high-throughput screening studies have targeted TDP-43 to identify its preferred RNA targets. These studies have been performed in variety of cell lines such as HeLa, 93 HEK293E,94,95 mouse Neuro2a cells, 96 induced pluripotent stem cells (iPSCs) derived from patients with ALS, 97 and transgenic mouse models overexpressing wild-type and mutant versions of human TDP-43. 98 In addition, immunoprecipitation analysis coupled with deep sequencing has also been recently used to identify TDP-43 mRNA targets in normal mouse brain 74 and in the cytoplasm of NSC-34 cells. 99 The aim of these studies was to identify disease-specific RNA processing events that become altered in disease and could therefore be rescued using the various methodologies that have been recently developed in the RNA processing field100,101 ( Fig. 2 ).
As with the protein-protein interaction section, the major problem in targeting RNA-protein interactions of TDP-43 is limited by our ability to exactly identify which events should be targeted among the several hundreds that are normally described to occur for any RBP of interest. As this particular topic has already been discussed elsewhere in detail, the reader is referred to the recent review by Buratti et al. 102
Nonetheless, just to give an example of the complexity of results when trying to address this issue, Table 3 provides an overview of the major RNA-protein interaction networks of TDP-43. In particular, TDP-43–dependent changes of gene expression have been functionally validated for five targets, and the mRNA regions targeted by this factor have been mapped. In addition, at least 25 additional alterations of TDP-43–dependent alternative splicing have been characterized so far. However, it should be noticed that only in 4 of 25 cases (CFTR, ApoAII, SC35, SKAR/POLDIP3, and SMN2), the regions required for these TDP-43–induced splicing variations have been found. Moreover, looking in deeper detail, the physical TDP-43–RNA interaction has been specifically mapped only in three genes (CFTR, SKAR/POLDIP3, and ApoAII), and in all the remaining cases, it has yet to be determined whether the involvement of TDP-43 is direct or indirect. For this reason, it is very likely that therapeutic interventions based on modulating the interaction of TDP-43 with specific RNAs are rather far in the future.
Validated TDP-43 RNA-Protein Interactions.
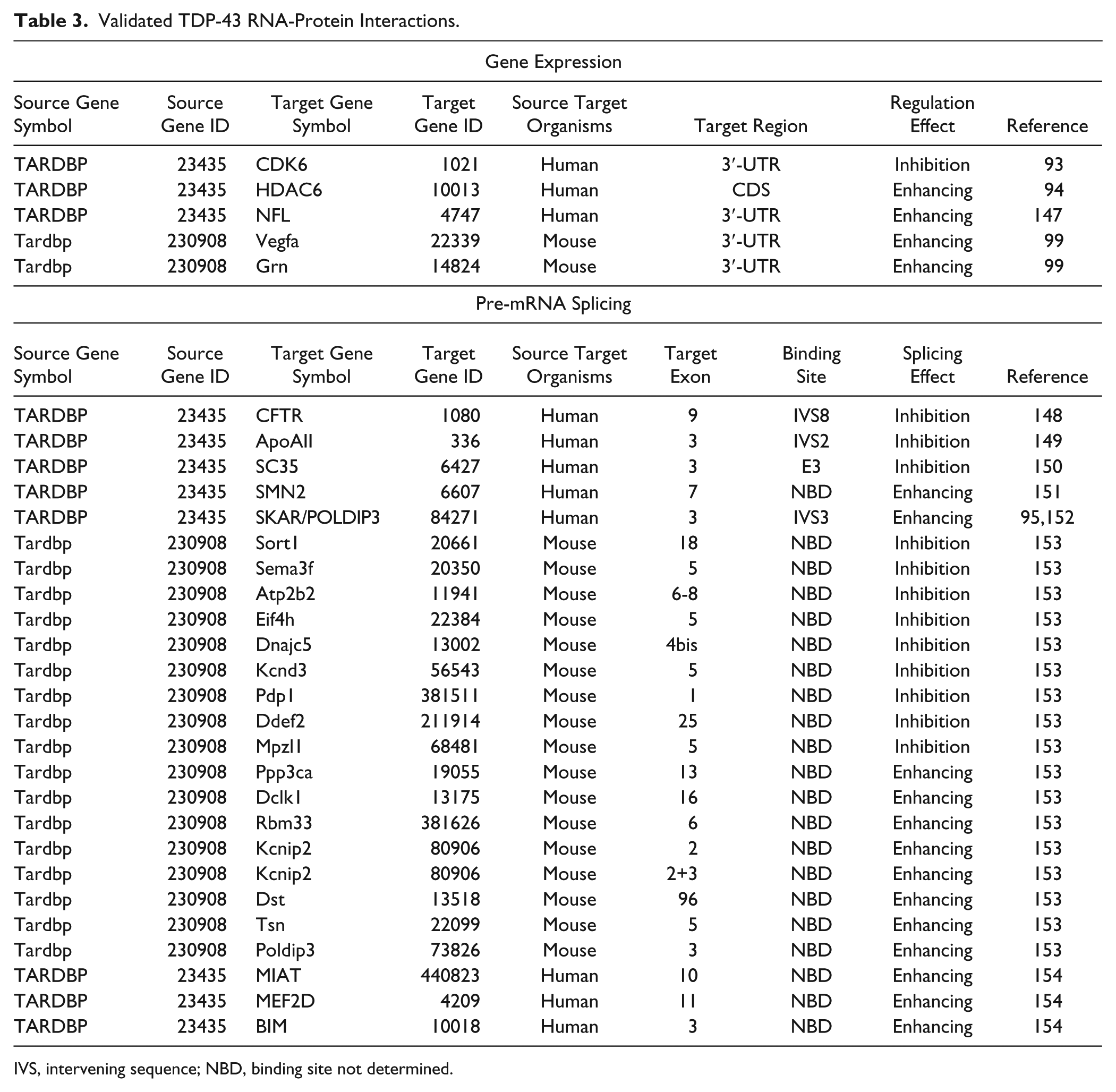
IVS, intervening sequence; NBD, binding site not determined.
The modulation of autoregulatory processes of RBPs might represent another potential therapeutic target. These processes often involve the presence of a negative feedback loop controlled by the binding of the RBP protein of interest to its own pre-mRNAs in the nucleus.11,103 Since these mechanisms are very efficient in raising/lowering protein production levels, any alteration in an RBP autoregulatory process may play an important role in a pathological setting, as already discussed by us elsewhere. 104 The reason is that pathological aggregates may act as a protein “sink” of the various RBP proteins, lowering their intracellular levels. This would predictably trigger an increasing rate of RBP production to overcome any “loss-of-function” effects in the nucleus, likely resulting in eventually toxic gain-of-function effects on the cellular metabolism. Even if successfully compensated, the increase in energetic expenditure to overcome the continuous loss of RBP from the nucleus may well lead to a certain degree of cell suffering if protracted for a long period. This could be especially harmful to neurons that have an extremely long life span and might help to explain why RBP alterations preferentially affect these types of cells.
Recently, autoregulation of TDP-43 expression levels has been shown to be dependent on its ability to bind several sequences 105 in the 3′-UTR region of its own mRNA. This region has been called TDPBR 106 and spans a normally silent intron that contains the major polyadenylation site (PAS) used by the TARDBP gene. Binding of TDP-43 to this region induces increased processing of the intron with the consequent use of suboptimal PAS sites that are retained in the nucleus and rapidly degraded. 107 As a result of this mechanism, a reduction of TDP-43 cellular mRNA levels can occur very rapidly in the presence of high TDP-43 concentrations.
From a biomolecular target point of view, therefore, one could hypothesize that these TDP-43 binding sequences in the 3′-UTR may represent a promising target to modulate the production of TDP-43 within cells, especially in a pathological setting to avoid the overexpression of this factor. Accessibility of these sequences by TDP-43 could thus be modulated using antisense oligonucleotides (AONs) 100 or other small RNA technologies 108 that are being developed just for this purpose. In particular, the development of high-throughput screening technologies for TDP-43 binding will be useful to identify compounds capable of modulating its binding to specific RNA targets. 109
Targeting TDP-43 Affected Pathways
One possible solution to overcome the overabundance of likely targets that originate from the very complex network of protein-protein and RNA-protein interactions described above might be to take a step backward and try to determine which cellular pathways are most affected following RBP dysregulation in disease.
Significant progress has been made in the neurodegeneration field using global techniques to identify cell signaling or metabolic networks involved in disease origin and progression, especially for ALS, AD, and PD, as recently reviewed elsewhere. 110 One of the major drawbacks of these approaches is that they often maintain a high degree of uncertainty with regard to the various candidates/hypotheses under investigation until the validation stage. Nonetheless, it is possible to reasonably guess which pathways may be preferentially affected following the aggregation/removal of each RBP factor.
Based on the protein-protein interactions reported in Table 1 for TDP-43, it is possible to identify the general pathways that might be affected if the protein becomes sequestered in the aggregates. In particular, Table 2 shows the SwissProt/PIR Protein Sequence Database keywords enriched in our set of TDP-43 protein-protein interactors. As reported in this table, 80% of the interactors were nuclear (p = 4.9 × 10−14) and associated with phosphoprotein (p = 1.6 × 10−7). More than 50% were associated with acetylation (p = 1 × 10−8), 26% with Ubl conjugation (p = 3 × 10−7), and 12% with the Ubl conjugation pathway (p = 0.02), respectively. In addition, 52%, 29%, 24%, and 19% were correlated with alternative splicing (p = 0.07), RNA binding (p = 1 × 10−8), mRNA processing (p = 3 × 10−9), and mRNA splicing (p = 3 × 10−7), respectively.
Taken together, this enrichment analysis supports the known functions of TDP-43 in the regulation of several steps of mRNA processing. In addition, if we consider its association with the ubiquitin pathway, one might be tempted to speculate that TDP-43 might have other still unmapped nonproteolysis-related roles such as, differentiation, inflammation, biogenesis of organelles, trafficking, and kinase activation.
Figure 3B
highlights in particular those pathways that are not necessarily the most prominent but might be targeted in a more profitable manner for specific therapeutic approaches (see options highlighted in
Future Aims
In conclusion, when studying the role of RBP proteins in neurodegenerative diseases, several major areas can be preferentially targeted to develop novel therapeutic or diagnostic approaches:
Aberrant aggregation processes of these factors represent the most prominent characteristic of disease. To maximize chances of success, priority should be given to obtain animal and cellular models that accurately mimic the pathology observed in patients. These models can then be used as ideal substrates to perform screening analysis to identify the genetic and environmental agents capable of modifying disease course and progression.
Additional targets can be identified through the characterization of the biological properties of each RBP factor of interest (and the way they become modified in the disease state). In particular, it is important to focus specific RNA processing events or protein-protein interactions that affect neuronal viability and synaptic functionality. In recent years, this approach has yielded several possible targets that are currently being validated. 111 In addition to these properties, this characterization should also involve a careful appraisal of how the RBP factor of interest relates to other factors or important disease modifiers. For example, the presence of nonpathological Atxn repeats may contribute to influence the onset and progression of disease in ALS and FTLD TDP-43 pathologies.112,113 The drawback of these approaches is that it is often very difficult to determine exactly which of the abundant RBP interactions plays a major role during disease.
An alternative to targeting specific events that are altered following aggregation could be to try and affect in a more general manner the pathways controlled by each individual factors. For example, molecules capable of generally improving the efficiency of autophagic or proteome clearance systems would certainly be beneficial in avoiding the build-up of the various aggregates.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by AriSLA (TARMA), Thierry Latran Foundation (REHNPALS), and University of Trieste-Finanziamento per Ricercatori di Ateneo.