
Abstract
The spliceosome is the macromolecular machine responsible for pre-mRNA splicing, an essential step in eukaryotic gene expression. During splicing, myriad subunits join and leave the spliceosome as it works on the pre-mRNA substrate. Strikingly, there are very few small molecules known to interact with the spliceosome. Splicing inhibitors are needed to capture transient spliceosome conformations and probe important functional components. Such compounds may also have chemotherapeutic applications, as links between splicing and cancer are increasingly uncovered. To identify new splicing inhibitors, we developed a high-throughput assay for in vitro splicing using a reverse transcription followed by quantitative PCR readout. In a pilot screen of 3080 compounds, we identified three small molecules that inhibit splicing in HeLa extract by interfering with different stages of human spliceosome assembly. Two of the compounds similarly affect spliceosomes in yeast extracts, suggesting selective targeting of conserved components. By examining related molecules, we identified chemical features required for the activity of two of the splicing inhibitors. In addition to verifying our assay procedure and paving the way to larger screens, these studies establish new compounds as chemical probes for investigating the splicing machinery.
Introduction
Pre-mRNA splicing is a critical process in eukaryotic gene expression. Although the chemistry behind removing introns and ligating exons is well understood, the mechanisms by which the spliceosome recognizes its substrates and regulates splicing remain unclear. Spliceosomes assemble de novo at introns in a stepwise process from five uridine-rich small nuclear RNAs with associated proteins (U1, U2, U4, U5, and U6 snRNPs) and a large number of additional protein components. 1 In vitro studies using native gels have defined an ordered series of intermediate splicing complexes. 2 In the first complex (E complex), U1 snRNP joins the pre-mRNA, followed by addition of U2 snRNP to create the prespliceosome or A complex. The U4, U5, and U6 tri-snRNP then join to create B complex, which is activated by release of U1 and U4 for splicing catalysis in C complex. Complex rearrangements of protein-protein, protein-RNA, and RNA-RNA interactions drive spliceosome assembly and progression. Given the complexity of the spliceosome, many additional complexes surely remain to be captured and characterized.
To make new intermediate spliceosome complexes available for biochemical and structural analysis, small-molecule inhibitors that selectively target different components are needed to arrest spliceosome progression at discrete steps. With the large number of enzymatic activities and regulated rearrangements in spliceosomes, it is clear that a diverse set of compounds will be required. Some splicing inhibitors may also be useful as biological probes of spliceosome function in cells. With the recent finding of spliceosome mutations associated with progression of chronic lymphocytic leukemia and myelodysplastic syndrome,3–6 such molecules may also hold promise for understanding and possibly treating human disease. 7
High-throughput screening (HTS) with a sensitive and robust assay is an important strategy for identifying small-molecule inhibitor candidates. An established human in vitro splicing system allows spliceosome function to be assessed in isolation from other cellular processes and provides a means to probe all of its ~100 components simultaneously.8,9 Here we describe HTS of ~3080 compounds for splicing inhibitors using a new reverse transcription followed by quantitative PCR (RT-qPCR) assay system. We identified three structurally distinct small molecules that inhibit human in vitro splicing reactions in a dose-dependent manner. We characterized the effects of these compounds on splicing chemistry and spliceosome assembly using extracts and substrates in human and yeast to examine their selectivity. One compound, tetrocarcin A (C1), an antibiotic with antitumor activity, 10 inhibits first-step chemistry at an early stage of spliceosome assembly in extracts from both organisms. A family of naphthazarin compounds (C3) affects later stages of spliceosome assembly in human and yeast extracts, whereas a third indole derivative (C2) blocks the earliest stages of assembly in the human system only. With these results, it is clear that we have an assay system that is robust in identifying new small-molecule modulators of splicing. Furthermore, we can attribute effects of candidate inhibitors to discrete steps of splicing chemistry and spliceosome assembly.
Materials and Methods
In Vitro Splicing Reactions
For the human splicing system, pre-mRNA substrate is derived from the adenovirus major late transcript. A G(5′)ppp(5′)G-capped substrate was generated by T7 runoff transcription followed by G50 gel filtration to remove unincorporated nucleoside triphosphates. Transcripts derived from a cDNA copy of spliced mRNA were used in some experiments as a control. For gel-based splicing assays, the substrate was body labeled with 32P-UTP. Nuclear extract was prepared from HeLa cells grown in MEM/F12 1:1 and 5% (v/v) newborn calf serum. 11 For splicing reactions, we incubated substrate RNA at 10 nM concentration in 60 mM potassium glutamate, 2 mM magnesium acetate, 2 mM ATP, 5 mM creatine phosphate, 0.05 mg mL−1 tRNA, and 50% (v/v) HeLa nuclear extract at 30 °C. For yeast splicing reactions, extracts were prepared according to Yan et al. 12 and assayed using RP51A pre-mRNA at 4 nM as previously described. 13
RT-qPCR reagents
RT-qPCR reactions were carried out using the TaqMan One-Step RT-PCR kit (Applied Biosystems, Foster City, CA) with the following primers and TaqMan probe: 5′-TCTCTT-CCGCATCGCTGTCT-3′ (forward primer) directed to the 5′ exon, 5′-GCGAAGAGTTTGTCCTCAACGT-3′ (reverse primer) directed to the 3′ exon, and 5′FAM-6-AGCTGTTG-GGCTGCAG SPC3-BH13′ (TaqMan probe) directed to the exon junction. We determined the qPCR efficiency for these primers as (10(–1/slope)–1), where slope was derived from the linear regression analysis from a standard curve of values for cDNA containing spliced mRNA.
High-Throughput Splicing Assay
In vitro splicing reactions were prepared in 384-well plates by dispensing 5 µL of nuclear extract by a liquid-handling robot (Janus; PerkinElmer, Waltham, MA). A second robot equipped with a 384-pin tool (Janus MDT; PerkinElmer) transferred 200 nL of library or control compounds dissolved in DMSO into the nuclear extract, then 5 µL splicing mix containing pre-mRNA substrate and buffer were added for final concentrations of 50% (v/v) nuclear extract, 200 µM library compound, 10 nM substrate RNA, 60 mM potassium glutamate, 2 mM magnesium acetate, 2 mM ATP, 5 mM creatine phosphate, and 0.05 mg mL–1 yeast tRNA. Plates were sealed and incubated for 60 min at 30 °C. After incubation, the splicing reaction was diluted 1:2 with water using a peristaltic dispenser (Matrix WellMate; Thermo Scientific, Waltham, MA).
RT-qPCR Analysis
A total of 15 nL of diluted splicing reaction was transferred by pin robot to a new 384-well plate containing 5 µL of RT-qPCR premix (1x TaqMan master mix, 0.8 µM reverse primer, 0.4 µM forward primer, 0.5 µM TaqMan probe). RT-qPCR plates were analyzed with an ABI PRISM 7900HT Sequence Detection System under the following conditions: RT-step: 30 min at 48 °C, 10 min at 95 °C; qPCR: 40 cycles of 30 s at 95 °C, 50 s at 50 °C, 50 s at 72 °C followed by 7 min at 72 °C. Threshold cycle values (CT) for individual wells were normalized on a plate-to-plate basis to uninhibited control reactions.
Z′ Calculation
We calculated a Z′ value for the assay using the following equation: Z′ = 1 − {(3σno splicing + 3σsplicing) /(µno splicing − µsplicing)}. “No splicing” values were derived from splicing reactions containing a pre-mRNA substrate with a mutation that blocks the second step of splicing chemistry, whereas normal “splicing” values were derived from splicing reactions with a pre-mRNA substrate competent for both steps of splicing chemistry.
Denaturing Gel Analysis
RNA was extracted from in vitro splicing reaction and separated on a 15% (v/v) denaturing polyacrylamide gel.32P-labeled RNA species were visualized by phosphorimaging and quantified with ImageQuant software (Molecular Dynamics, Sunnyvale, CA). Splicing efficiency is calculated as the amount of spliced mRNA relative to total RNA and normalized to a DMSO control reaction. IC50 is the concentration of inhibitor that causes 50% decrease of splicing efficiency estimated from plots of splicing efficiency versus compound concentration.
Native Gel Analysis
For human splicing complexes, nuclear extract was preincubated at 30 °C for 15 min to deplete endogenous ATP before setting up in vitro splicing reactions. Time point samples were kept on ice until all samples were ready for analysis. Ten microliters of splicing reactions were mixed with 5 µL native gel loading buffer (20 mM Trizma base, 20 mM glycine, 25% (v/v) glycerol, 0.05% (w/v) cyan blue, 0.05% (w/v) bromophenol blue, 2.5 mg mL–1 heparin sulfate) and incubated at room temperature for 5 min before loading onto a 2.1% (w/v) agarose gel. Gels were run at 72 V for 3.5 h, dried onto Whatman paper, and exposed to phosphorimaging screens, which were digitized with a Typhoon Scanner (Molecular Dynamics).
For yeast splicing complexes, 5 µL of yeast splicing reactions were mixed with 5 µL complex buffer, and yeast splicing complexes were separated using 0.5% (w/v) agarose; 3% 80:1 acrylamide:bis (v/v) gel run for 16 h at 8 5V. 13
Results
RT-qPCR Assay to Screen for Inhibitors of In Vitro Splicing
To search for inhibitors of the human spliceosome, we used a synthetic pre-mRNA substrate consisting of two exons separated by an intron and HeLa nuclear “splicing” extract.
8
Spliceosomes assemble on the pre-mRNAs, which are then spliced in two chemical steps forming mRNA9,14 (
Fig. 1a
). Typically, the reaction is analyzed by denaturing gel electrophoresis to quantify the amount of splicing products, which is not conducive to scale up or automation. For HTS, we developed an RT-qPCR assay that employs a TaqMan probe complementary to the unique splice junction sequence created by exon ligation (
Fig. 1b
). This assay reports a threshold cycle (CT) that directly correlates to the amount of mRNA produced in the reaction by the spliceosome. A lower CT value indicates more mRNA and more splicing, whereas an increased CT value corresponds to reduced mRNA and less splicing. Using this system, we readily detect mRNA produced by in vitro splicing (
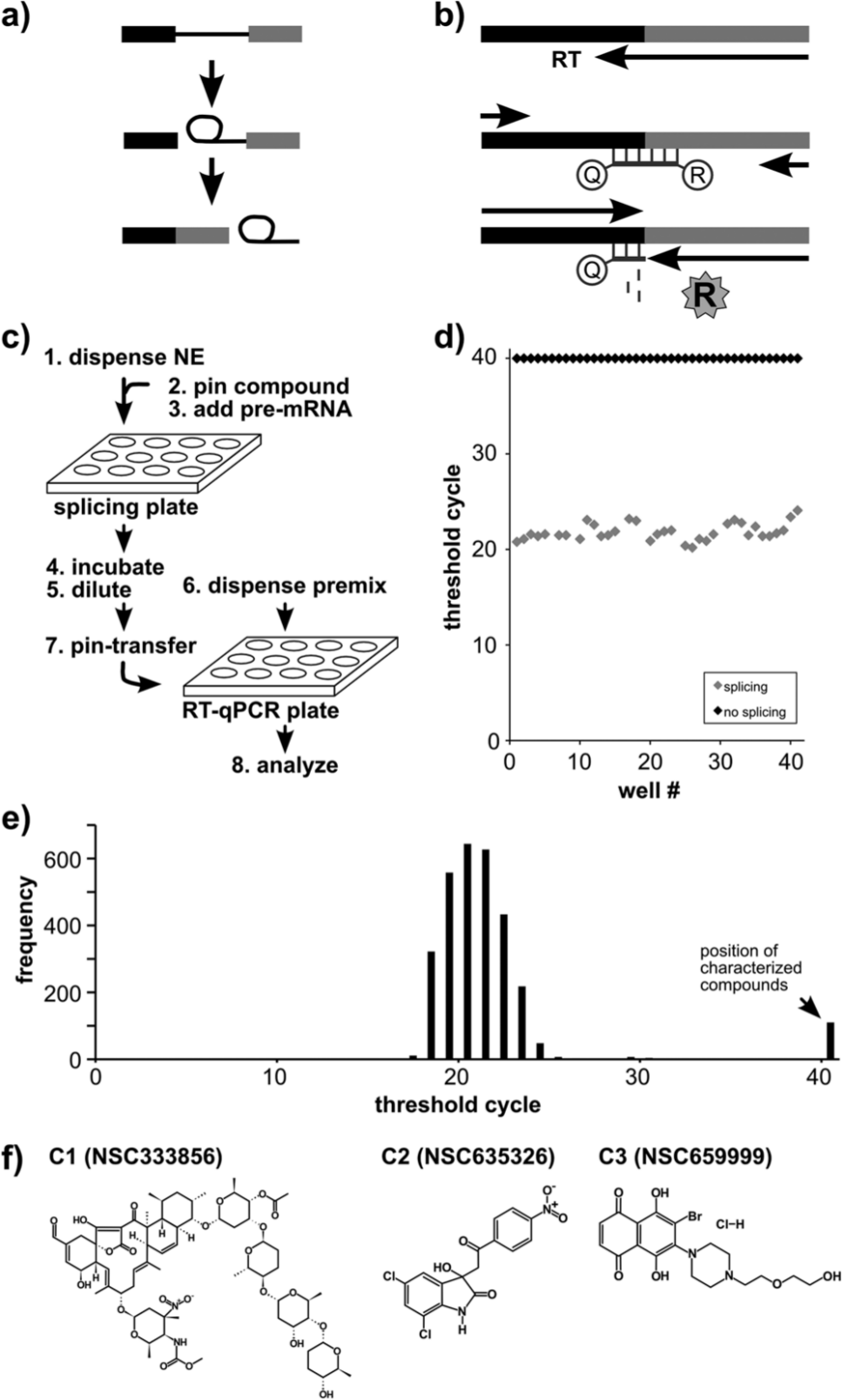
High-throughput approach to identify splicing inhibitors. (
To screen large chemical libraries for splicing inhibitors, we used liquid-handling robots to set up the assay in 384-well plates. The steps of the protocol are schematized in Figure 1c : (1) 5 µL nuclear extract is dispensed to wells, (2) a pin robot transfers 0.2 µL of test compounds from a library plate into the nuclear extract, (3) 5 µL of pre-mRNA substrate in splicing buffer is dispensed into the nuclear extract, (4) splicing proceeds for 60 min, (5) the reactions are diluted with 10 µL of water, (6) 5 µL of the RT-qPCR reaction components are dispensed into a second 384-well plate, (7) a pin robot transfers 15 nL of the diluted splicing reactions as a template to the RT-qPCR plate, and (8) RT-qPCR is carried out. To analyze the data, we compare the CT value for each well with control splicing wells into which either DMSO alone or SSA was added.
In scaling up for screening large compound libraries, some assay steps can be performed ahead of time. For example, we prefill plates with nuclear extract, which is labile to some extent, and store frozen to avoid extended waiting prior to setting up the splicing reaction. For convenience, we also prefill and freeze plates with the RT-qPCR reaction mix. Because the RT-qPCR analysis requires 3.5 h for each plate, we seal and freeze the PCR plates after pinning from the diluted splicing reaction and perform the analysis over several days as needed. A second practical matter is generating large quantities of nuclear extract with reasonable splicing capacity. Our lab currently grows HeLa cells in 10 L batches and prepares enough extract per month to screen 10,000 compounds. If desired, this capacity could be readily increased. Notably, we find that splicing efficiency varies with the source of the HeLa cell line (data not shown), so it is important to test extracts before using them for splicing reactions.
Before screening chemical libraries for splicing inhibitors, we characterized the splicing assay system by setting up a plate with alternating rows of reactions containing either wild-type pre-mRNA (normal splicing) or with pre-mRNA containing a splice site mutation that prevents the second step of splicing chemistry (no-splicing control). We measured mRNA amounts by RT-qPCR for each condition and found that normal splicing reactions cluster with an average CT value of 22 ± 1.07, whereas the no-splicing control reactions never reach threshold (>40 CT), as expected for no-spliced mRNA product ( Fig. 1d ). From these CT distributions, we calculate a Z′ value 16 for the assay of 0.82 with good separation (~18 CT) between fully normal and no-splicing splicing reactions. We conclude that the automated splicing assay can measure loss of splicing, allowing us to screen large chemical libraries for splicing inhibitors.
HTS for Splicing Inhibitors
Using the RT-qPCR splicing assay, we screened a 3080-compound library combining the Structural Diversity Set, Challenge Set, Natural Products Set, and Mechanistic Diversity Set from the National Cancer Institute (NCI). The vast majority of compounds in the library did not have an effect on in vitro splicing and showed low CT values within a baseline range centered at a CT value of 21 ( Fig. 1e ). About 3%, or 100 compounds, resulted in CT values in the fully inhibited splicing range ( Fig. 1e ; CT > 40). For these, we returned to the original splicing plate and manually repeated the RT-qPCR analysis of the candidate inhibitor wells in triplicate, which reconfirmed 40 (1.3% overall) as yielding high CT values. We ordered the compounds present in those wells and retested their effects on in vitro splicing by the same RT-qPCR assay. Six of these consistently interfered with in vitro splicing, but only three showed clear dose-dependent effects that are expected for a specific splicing inhibitor ( Fig. 1f ).
Candidate Compounds Inhibit Human and Yeast Pre-mRNA Splicing
The three candidate splicing inhibitors that we identified from the pilot screen have very distinct structures ( Fig. 1f ). Compound 1 (C1) is a large and complex natural compound known as tetrocarcin A (NSC333856). It has been previously described as an antibiotic 10 and as an antitumor compound that inhibits the anti-apoptotic gene Bcl2.10,17,18 The second compound (C2) is an indole derivative (NSC635326) with no known biological activity. The third compound (C3) is a naphthazarin derivative (NSC659999), and like several other naphthazarin compounds, it has shown activity in a variety of biological contexts, including suppression of tumor growth. 18
The RT-qPCR splicing assay detects inhibition of mRNA accumulation but does not identify the molecular basis of this inhibition. To determine how the three compounds affect splicing chemistry, we performed in vitro splicing assays in HeLa nuclear extract using radiolabeled pre-mRNA substrate followed by gel electrophoresis to visualize splicing intermediates and products. With all three compounds, unspliced pre-mRNA substrate levels did not change significantly, which indicates that RNA is generally stable in the reactions in the presence of compound. Therefore, RNA degradation induced by the compounds does not account for the absence of mRNA. Instead, the addition of all three compounds to splicing reactions results in a loss of splicing chemistry in a concentration-dependent manner (
Fig. 2a
). The presence of C1 primarily resulted in loss of the first step of splicing chemistry (and thereby second-step chemistry as well). At 100 µM of C1, all splicing chemistry disappeared, and we determined an IC50 for the compound of ~25 µM. Second-step chemistry appeared more sensitive to the C2 and C3 compounds, although first-step chemistry was also lost at higher concentrations (
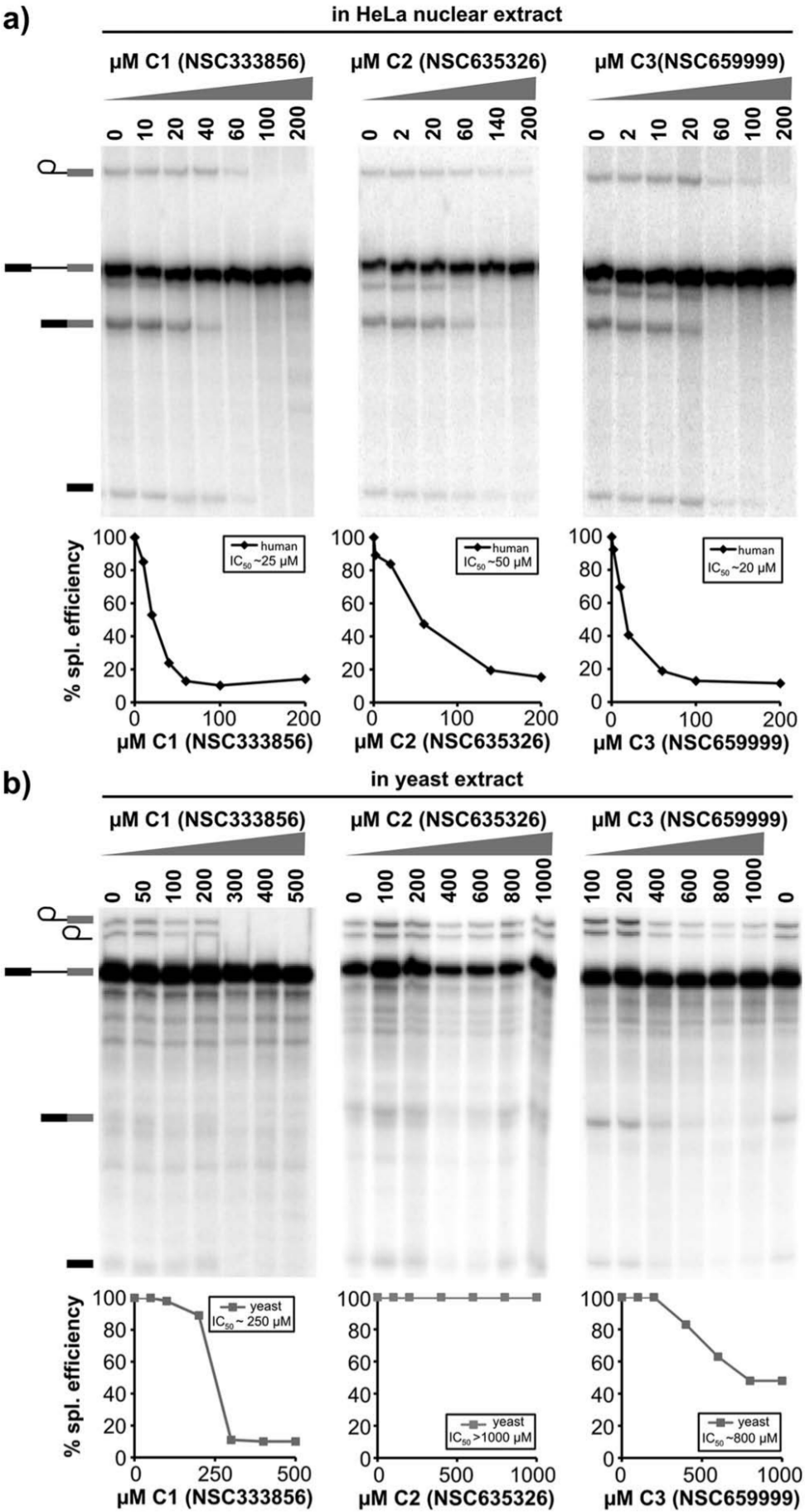
Three compounds inhibit splicing chemistry in a dose-dependent manner. Denaturing gel analysis of RNA from splicing reactions with an indicated concentration of C1, C2, and C3. Quantification of splicing efficiency versus inhibitor concentration is plotted below each gel along with estimated IC50 values. (
We also examined the effects of these compounds on in vitro splicing in Saccharomyces cerevisiae extracts with an RP51A splicing substrate 19 ( Fig. 2b ). Again, addition of C1 resulted in loss of first-step chemistry, although higher concentrations were required to see an effect with an IC50 of ~250 µM. Similar to its effect in human extracts, addition of C3 primarily resulted in loss of second step, although much higher concentrations are required and splicing is never completely blocked. In contrast, C2 had no effect on yeast splicing. These results suggest that C1 and C3 interfere with a conserved mechanism of the splicing process, whereas C2 is selective for a factor specific to human splicing and does not simply inactivate all splicing extracts.
C1 Stalls Spliceosome Assembly Early in Human and Yeast Extracts
The spliceosome assembles through a series of complex intermediates, and we expect that a splicing inhibitor selective for a component involved in complex formation will interfere with a specific assembly stage. We used native agarose gels to investigate the effect of inhibitor compounds on human spliceosome assembly. 20 In the absence of inhibitor (2% DMSO), these gels show the progression of spliceosome assembly through complex intermediates of E/H, A, B, and C complexes ( Fig. 3b , lanes 1–5). Both steps of splicing chemistry take place in C complex, which disassembles immediately after catalysis is completed. Direct inhibition of splicing chemistry typically causes C complex accumulation, whereas interference with a specific assembly step often results in accumulation of the preceding complex.
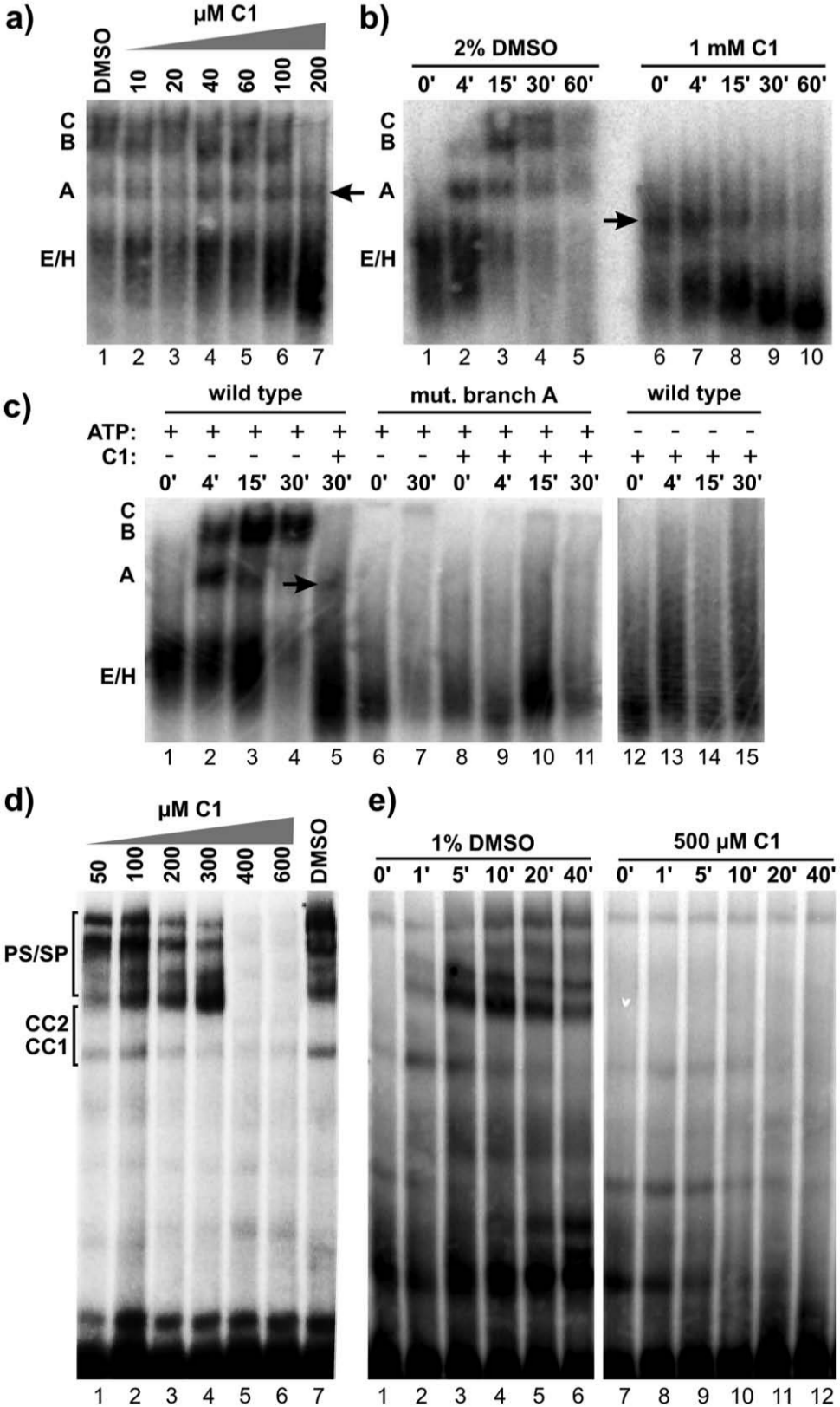
C1 stalls human and yeast spliceosomes at an A-like complex. Native gel analysis of spliceosome assembly. (
In the presence of increasing amounts of C1, spliceosome assembly is lost in parallel with the loss in splicing chemistry ( Figs. 3a and 2a ). At a concentration that completely blocks splicing, a complex that migrates near the position of A complex (“A-like” complex) accumulates. Furthermore, B and C complexes do not appear over time, but instead an apparent conversion of the A-like complex to a stable faster migrating species becomes evident ( Fig. 3b , lanes 6–10).
Formation of A complex requires recognition of the branch point sequence and ATP hydrolysis.2,21 If the A-like complex is related to normal spliceosome assembly, it should have the same substrate and ATP requirements. Splicing reactions assembled in the presence of C1 with a pre-mRNA mutated at the branch point sequence no longer produce the A-like complex ( Fig. 3c , lanes 8–11). Like a branchpoint in the substrate, ATP is also required to form the A-like complex ( Fig. 3c , lanes 12–15). This result shows that compound C1 does not generally disturb all components of the extract, because specific dependencies of spliceosome assembly are maintained. It also suggests that C1 interferes with a component(s) involved in the stability of A complex or the transition to the next assembly stage.
We also looked at spliceosome assembly in yeast extracts treated with C1 and again see a dose-dependent loss of spliceosome assembly compared with DMSO alone ( Fig. 3d ). At lower concentrations, the effect of C1 is most evident in the appearance of prespliceosomes/spliceosomes (PS/SP) complex bands, which are equivalent to A/B/C complexes in human extracts ( Fig. 3D , lanes 1–4), whereas at higher concentrations, all PS/SP accumulation is lost (lanes 5–6). At 500 µM concentration, C1 also dramatically reduces stable accumulation of commitment complexes CC1 and 2, which are formally equivalent to E/H complex in human extracts ( Fig. 3e , lanes 7–12).
From the yeast and human data, we conclude that C1 does not directly inhibit first splicing chemistry but instead interferes with early stages of spliceosome assembly in both systems potentially by destabilizing complexes that form. These results also suggest that it targets a core component conserved in both human and yeast spliceosomes.
C3 Causes accumulation of a B-like Complex
C3 had a different effect on spliceosome assembly in human extracts. As the amount of C3 is increased in splicing reactions, C complex decreases with the same dose dependence as loss of second-step chemistry ( Fig. 4a , lanes 2–7, and Fig. 2a ). The compound does not inhibit or change the timing of A and B complex formation, even at concentrations that completely block both steps of splicing chemistry ( Fig. 4a , lanes 13–17). This result shows that C3 does not generally disrupt complex assembly. Instead, considering the different sensitivities of first- and second-step chemistry to C3, there must be a factor(s) involved in late assembly that is selectively targeted by the presence of the compound.
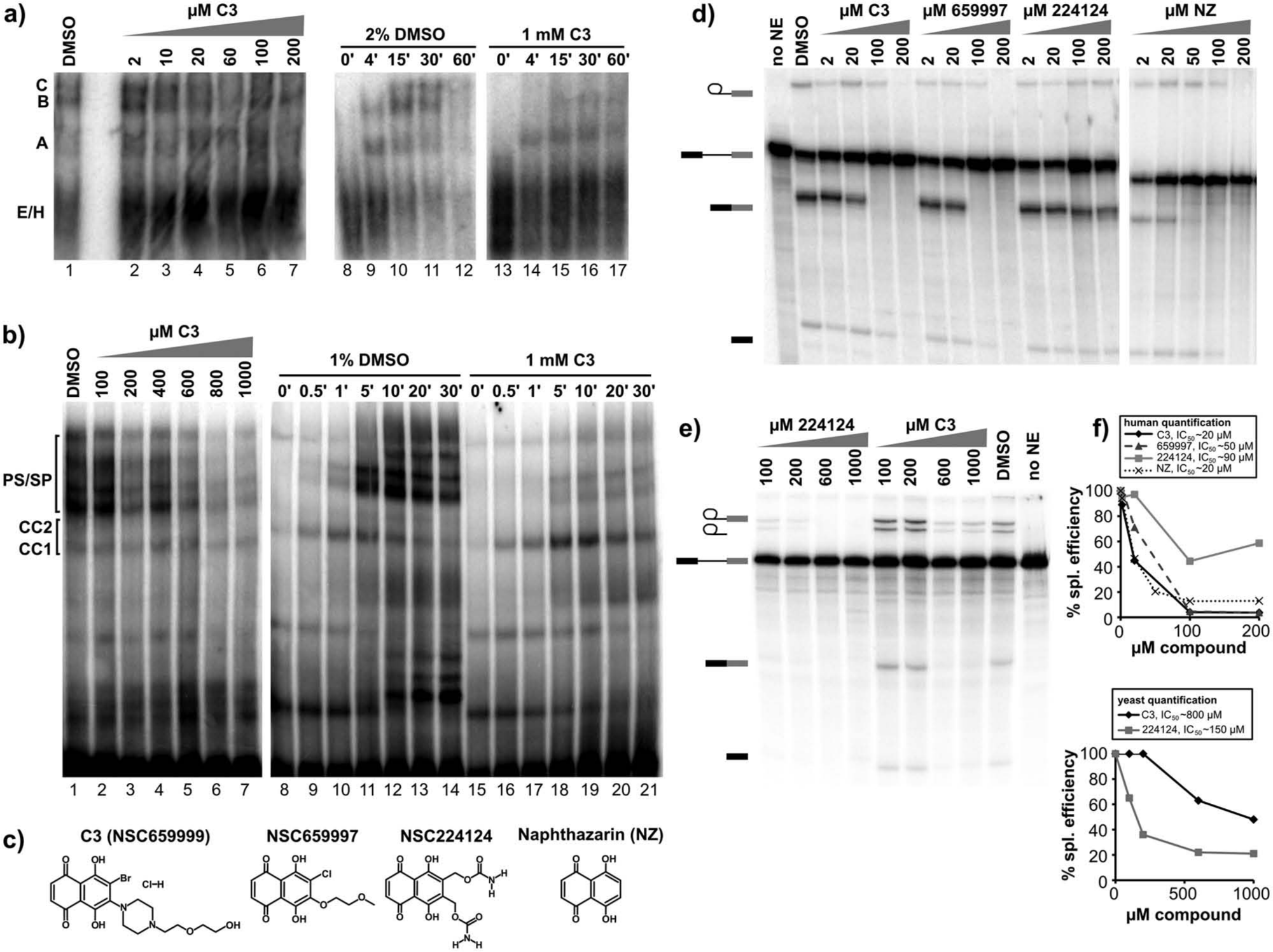
A B-like complex accumulates in the presence of the C3 compound. (
In yeast extracts, there is a similar dose-dependent loss of spliceosome assembly ( Fig. 4b , lanes 2–7) with C3. In comparison to no inhibitor (1% DMSO, lanes 8–14), progression from commitment complexes (CC1/CC2) to prespliceosomes/spliceosomes (PS/SP) is decreased ( Fig. 4b , lanes 15–21), which is formally equivalent to a decrease in A/B/C complex formation in human extracts.
C3 Activity Is Related to Redox Potential in HeLa Extract
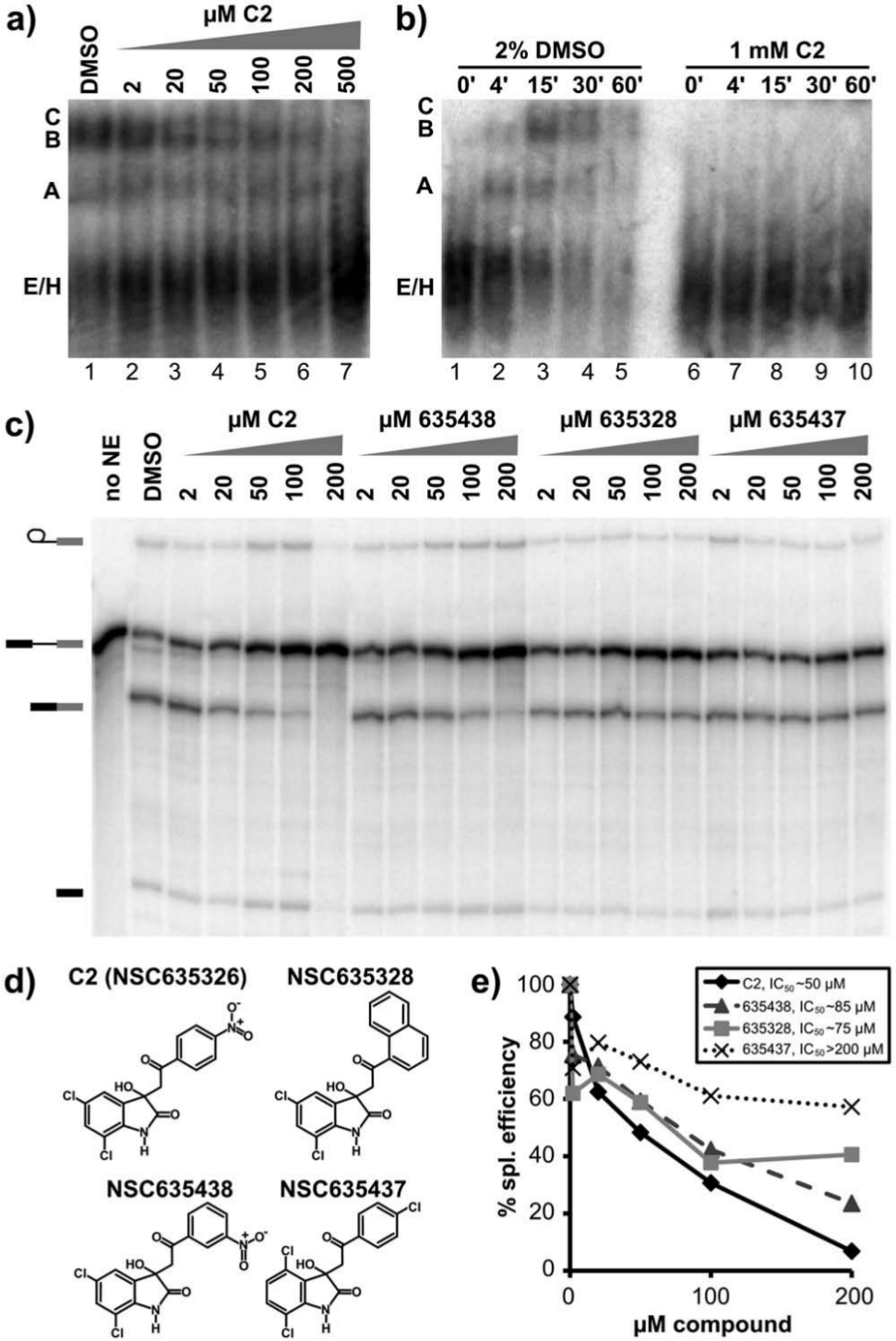
To examine the structure-activity relationships of the C3 inhibitor, which contains a naphthazarin backbone, we tested the effects of related compounds on in vitro splicing. In human extracts, another naphthazarin derivative (NSC659997) inhibits splicing primarily at second step, although with a slightly higher IC50 of ~50 µM ( Fig. 4c , 4d , and 4f ). Interestingly, NSC659997 also shows growth inhibition in a panel of tumor cell lines that is similar to the original C3 compound. 18 A second naphthazarin derivative (NSC224124) had a more limited effect on splicing ( Fig. 4c , 4d , and 4f ), and no effect on same panel of tumor cells. Finally, we tested the naphthazarin backbone alone (NZ) and find that it also inhibits splicing with an IC50 similar to C3 ( Fig. 4c , 4d , and 4f ). Together, these results suggest that the naphthazarin backbone alone mediates splicing inhibition but that substitutions within the ring structure can affect the activity.
These results are in some contrast to what we observe with naphthazarin derivatives in yeast splicing. In this case, the naphthazarin backbone alone was not sufficient to inhibit splicing chemistry (data not shown). In contrast, NCS224124 is more potent in yeast than the C3 compound but still has a relatively high IC50 of ~150 µM ( Fig. 4e and 4f ). This difference in activity points to the importance of substitutions on the naphthazarin ring in yeast splicing as well, but it is not clear why the sensitivity to the substitutions is different for human splicing.
Naphthazarin and many of its derivatives are reactive compounds with well-known redox properties proposed to interfere with proteins by two mechanisms. 22 First, they can serve as electrophiles to covalently modify proteins, most commonly by thioether linkage with labile cysteine residues, which cannot reverse by DTT. Alternatively, they also generate reactive oxygen species (ROS) that, among other effects, can oxidize thiol groups of cysteines, a process that can be blocked by addition of DTT. We tested the effect of DTT on HeLa splicing and found that excess DTT recovers about 70% of splicing in reactions inhibited by the naphthazarin compounds ( Fig. 5a and 5b ). DTT alone does not affect splicing efficiency up to 20 mM concentration. Relief of splicing inhibition by DTT suggests that the compounds inhibit splicing through ROS generation that results in modification of a redox-sensitive cysteine(s). Because we still see assembly to B complex in the presence of the naphthazarin compounds at all concentrations, the redox-sensitive cysteine is specific and appears to be required for formation of a catalytically active spliceosome. Surprisingly, in yeast extracts, DTT did not reverse splicing inhibition by the most potent naphthazarin derivative (NCS224124; Fig. 5c and 5d ), which suggests that the compound functions by a different mechanism in yeast splicing. Again, we cannot specify why different ring substitutions in the naphthazarin backbone affect the inhibitory properties of the different compounds. With human splicing, they may modulate the redox potential of the compounds, whereas splicing in yeast appears sensitive to their structure.
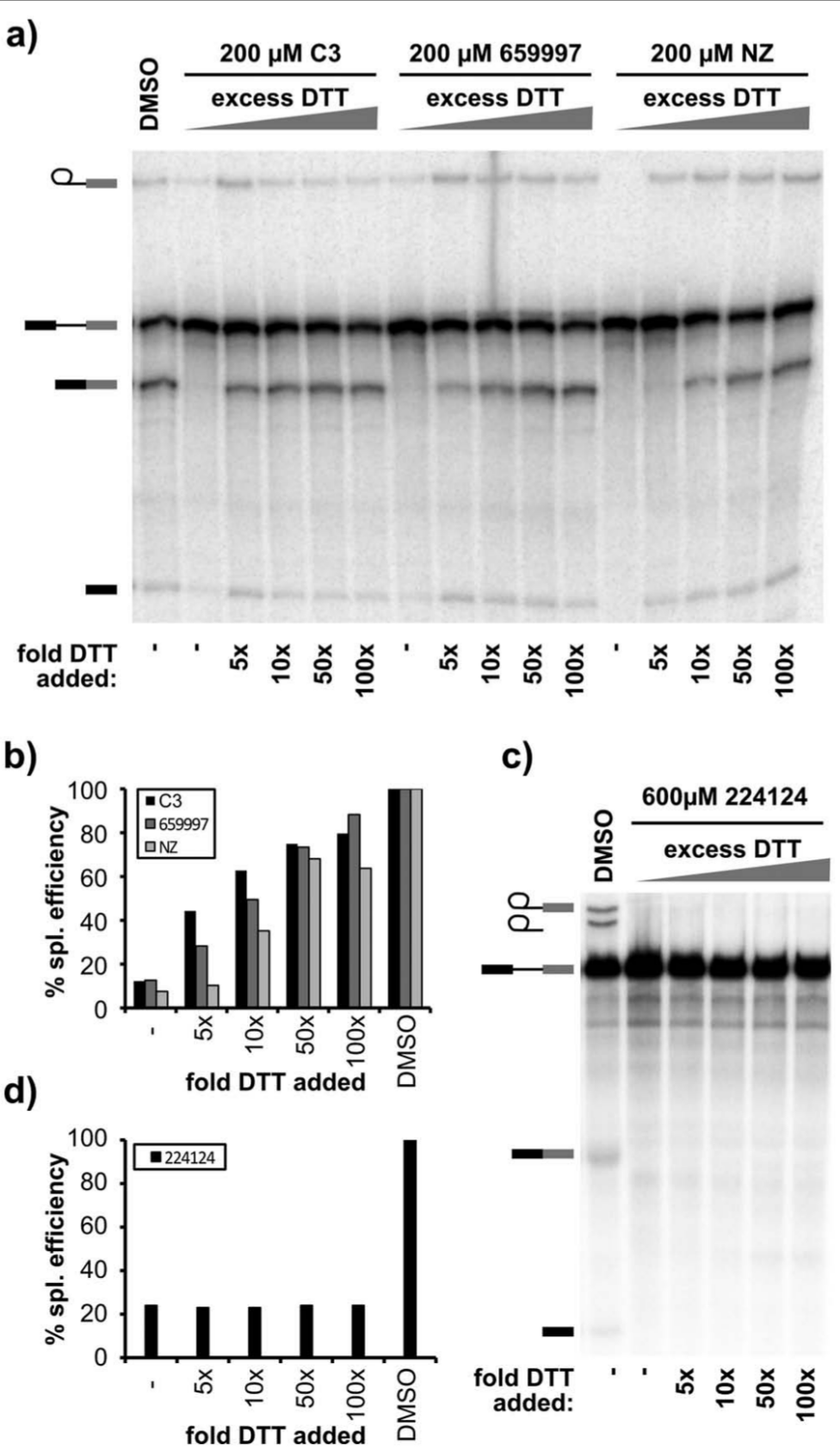
Inhibition by naphthazarins is partially rescued by excess DTT. (
C2 Inhibits Spliceosome Assembly Early in Human Extracts
The C2 compound only inhibits in the human splicing system and primarily affects very early spliceosome assembly. With increasing concentrations of C2, there is a loss of higher complexes that correlates with the loss of splicing chemistry (
Figs. 6a
,
2a
, and
Presence and orientation of a nitrophenyl ring are important for splicing inhibition by C2. (
We also examined structure-activity relationships for C2. Two related compounds with different substitutions in place of the nitrophenyl group had very little effect on splicing chemistry ( Fig. 6c , 6d , and 6e ). A third compound, which differs only in the linkage position of the nitrophenyl group, inhibits splicing but with less potency than the original C2 compound ( Fig. 6c , 6d , and 6e ). We conclude that the presence and orientation of a nitrophenyl ring from the oxoethyl group at the 3′ position of the indole ring structure are important for the mechanism of C2 inhibition of splicing. Possible explanations for the requirement include specific spliceosome component interaction with the nitrophenyl ring, increased stability of the compound in extracts, or increased solubility.
Discussion
Using a highly sensitive RT-qPCR splicing assay, we identified three structurally diverse molecules (C1, C2, and C3) that inhibit pre-mRNA splicing at specific stages in spliceosome assembly in both HeLa and S. cerevisiae extracts. In HeLa extracts, this inhibition has an IC50 in the 20 to 50 µM range. These three compounds are structurally distinct from previously identified splicing inhibitors23–25 and offer a unique opportunity to dissect important spliceosomal complexes not previously captured in either human or yeast splicing systems.
C1, also known as tetrocarcin A, has demonstrated antibiotic activity as well as cytotoxic effects in several cancer cell lines. 10 It has been shown to promote apoptosis by blocking BCL2 activity, 17 activating caspase-9, 26 or inhibiting PI3K kinase activity, 27 depending on cell type, although its molecular mechanisms are not clear. One possibility for this wide range of effects is that tetrocarcin A inhibits the splicing of one or more key gene products in the apoptosis pathway.
C2 impedes all stages of spliceosome assembly, eventually resulting in loss of all complexes beyond E/H. C2 has no effect on yeast in vitro splicing; therefore, its target(s) are likely specific to higher eukaryotes. By comparing the activity of C2 to several structurally related compounds, we find that a nitrophenyl substituent is key to its inhibition of splicing. The lack of previous data demonstrating an effect of C2 on cell growth suggests cells may not readily take up the compound. However, it still may have in vitro utility by allowing concentration-dependent access to the earliest stages of spliceosome assembly.
The influence of C3 (NSC659999) on splicing is more complicated. At lower concentrations, this compound primarily affects second-step chemistry, which is mirrored by a clear loss of C complex accumulation. In yeast, C3 shows similar effects on splicing chemistry and complex assembly, although only at significantly higher concentration. In the human system, the effect of the C3 and related naphthazarin inhibitors can be reversed by the addition of excess DTT, which suggests that ROS generation by these compounds plays a role in their inhibition of splicing. It also indicates that a specific redox-sensitive cysteine(s) in a splicing component functions in the transition from first- to second-step chemistry. Recently, the Dreyfuss lab identified a CDC25 phosphatase inhibitor in their screen for compounds that block in vitro splicing. 24 This compound is a naphthoquinone (NSC95397) with redox properties similar to the napthazarins. It also affects second-step chemistry and C complex formation such as the naphthazarins. It is notable that the phosphatase PP2A is also required for second-step chemistry in human extracts and that the PP2A inhibitor okadaic acid is also a splicing inhibitor.28,29 PP2A is sensitive to oxidation by H2O2, which can also be rescued by excess DTT. 30 We propose that at least some of the splicing inhibition observed with naphthoquinones, which include the naphthazarins, is conferred by indirect inactivation of PP2A by oxidation. As of yet, there is no known role in splicing for the yeast PP2A ortholog. This may explain why most of the naphthazarin derivatives had no effect on yeast splicing and why the modest effect of NSC2241124 in yeast extracts cannot be rescued by DTT.
There are several directions of study that may be pursued with these new splicing inhibitors. Our results underscore their potential as tools in vitro to study splicing mechanisms. For those that can be synthesized and derivatized, their structure-activity relationships can be further explored and, in some cases, their targets potentially identified by addition of affinity tags. These studies may also open the door to their use as probes in studying the role of splicing in cells. Many splicing inhibitors have shown bioactivity in the growth of different tumor cell lines, including tetrocarcin A and naphthazarins.17,22,31,32 An important next step will be to determine whether this activity is due to inhibition of spliceosomes and, if so, which splicing pathways are particularly affected.
Finally, with the extreme complexity of the spliceosome, the need is still great for a larger arsenal of compounds that modulate enzymatic functions and rearranging interactions involved in splicing. Fortunately, a great deal of chemical space remains unexplored. So far, only relatively small libraries of bioactive molecules (<8000 compounds) and one larger library of synthetic small molecules (30,000 compounds) have been screened.23–25 As we use the assay presented in this article to screen more compound libraries, particularly those containing structurally diverse natural products, we will certainly increase the number of small-molecule tools that will be useful for studying splicing mechanisms and cellular functions.
Footnotes
Acknowledgements
We thank the UCSC Chemical Screening Center, supported by NIH 1S10RR022455 and grants from the California Institute for Quantitative Biosciences (QB3) and the U.S. Department of State. We also thank J. Woo at the UCSF Genomics Core Facility for assistance with RT-qPCR analysis, M. Yoshida at the RIKEN Advanced Science Institute for providing SSA, B. Prichard for assistance in HeLa cultures and extract preparation, and N. Weber for developing scripts to help analyze RT-qPCR results.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the University of California Cancer Research Coordinating Committee and National Institutes of Health grant R01CA136762 to M.S.J. We thank the UCSC Chemical Screening Center, supported by NIH 1S10RR022455 and grants from the California Institute for Quantitative Biosciences (QB3) and the US Department of State.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.