
Abstract
Baeyer-Villiger monooxygenases (BVMOs) have been receiving increasing attention as enzymes useful for biocatalytic applications. Industrial requirements call for rapid and extensive redesign of these enzymes. In response to the need for screening large libraries of BVMO mutants, we established a generic screening method that allows screening of Escherichia coli cells expressing active BVMOs in 96-well plate format. For this, we first developed an expression system for production of phenylacetone monooxygenase (PAMO) in the periplasm of E. coli. This allows probing the enzyme for any target substrate while it is also compatible with extracellular coenzyme regeneration. For coenzyme regeneration, we used phosphite dehydrogenase, which forms phosphate upon NADPH recycling. This allowed the use of a chromogenic molybdate-based phosphate determination assay. The screening procedure was supplemented with a detection method for identification of mutant enzymes that act as NADPH oxidases, thereby excluding false-positives. The whole-cell–based screening method was validated by screening site–saturation libraries of PAMO and resulted in the identification of PAMO mutants with altered catalytic properties. This new method can be used for screening libraries of BVMOs for activity with any desired substrate and therefore is a powerful tool for engineering of these enzymes.
Introduction
Baeyer-Villiger monooxygenases (BVMOs) are promising enzymes with respect to industrial applications as they offer the possibility to perform, among others, oxygen atom insertion into a C-C bond and enantioselective sulfoxidations.1,2 However, the properties of naturally occurring enzymes are typically far from the demands set by commercial processes. Features such as substrate acceptance, stereoselectivity, and stability need to be improved before these enzymes can be applied on an industrial scale. Directed evolution is a powerful tool for engineering enzymes. Despite the potential significance of BVMOs, there have been only a few examples of directed evolution experiments on this class of enzymes. In most of the reported studies, screening for BVMO variants with improved properties was performed using chromatographic methods such as gas chromatography (GC)3–7 or high-performance liquid chromatography. 8 Unfortunately, these screening methods are of limited throughput, demand establishing optimal separation conditions for each targeted compound, and require specialized equipment. In addition, the adrenalin assay was applied in screening for enantioselective variants of a BVMO from Pseudomonas fluorescens DSM 50106. 9 In this assay, the hydrolysis of Baeyer-Villiger products yielded 1,2-diols, which were then quantified in a colorimetric assay. Furthermore, Bornscheuer and coworkers used p-nitroacetophenone to detect 4-hydroxyacetophenone monooxygenase activity. 10 In this assay, Baeyer-Villiger oxidation of the substrate and hydrolysis of the product led to the formation of p-nitrophenolate. The assay was applied in screening libraries of 4-hydroxyacetophenone monooxygenase mutants yielding variants with improved activity toward p-nitroacetophenone. However, the latter 2 methods can only be used for specific substrates.
A few other methods for screening BVMOs have been described, for example, a pH change–based assay. 11 This assay revolves around hydrolysis of the Baeyer-Villiger product, resulting in the release of acid. However, this assay requires extensive optimization to give reliable signals and an esterase specific for the Baeyer-Villiger product. Further-more, several fluorogenic methods, employing umbelliferone-labeled substrates, have been used successfully for the detection of BVMOs.12,13 The underlying principle of the latter methods is the Baeyer-Villiger oxidation of the umbelliferone-labeled substrate and subsequent hydrolysis of the Baeyer-Villiger product, resulting in the release of the fluorescent umbelliferone. A screening method for ketone biotransformations was also presented, which was used successfully for the detection of BVMO activity. 14 This method is based on the reaction between alicyclic ketones and 3,5-dinitrobenzoic acid under alkaline conditions, resulting in the formation of a purple-colored product. Despite being fast and simple, this method requires relatively high activities, and it is limited to a certain class of compounds.
Although a variety of screening methods for the detection of BVMO activity has been described (see above), most of these methods have never been tested in BVMO mutant library screening, are of limited throughput, are designed for specific substrates, or require the synthesis of labeled substrates. The drawbacks of the current screening methods underscore the need of a generic, preferably high-throughput, screening method for BVMO activity. Here we report on a screening method for NAD(P)-dependent enzymes, in particular BVMOs ( Fig. 1 ). This screening system was validated for phenylacetone monooxygenase from Thermobifida fusca (PAMO, EC 1.14.13.92). 15 We expressed PAMO in the periplasm of Escherichia coli to ensure the unlimited access of substrates to the enzyme and to enable the shuttling of nicotinamide coenzymes between PAMO and externally added phosphite dehydrogenase (PTDH). The latter enzyme is required for NADPH regeneration. 16 Coenzyme regeneration by PTDH results in phosphate as a side product, which can be used as a reporter of BVMO activity. To quantify phosphate, we used a modified version of the molybdate assay, 17 which allows colorimetric detection. To increase the fidelity of our screening procedure, we included a control reaction to exclude false-positive clones. To this end, we detect hydrogen peroxide, which is typically produced during uncoupling of the BVMO reaction at the expense of NADPH and oxygen. 18 The integration of periplasmic expression of PAMO, coenzyme regeneration, and spectrophotometric detection of coupled products resulted in an accurate and reliable screening method, which can be applied to test monooxygenase activity on any desired substrate.
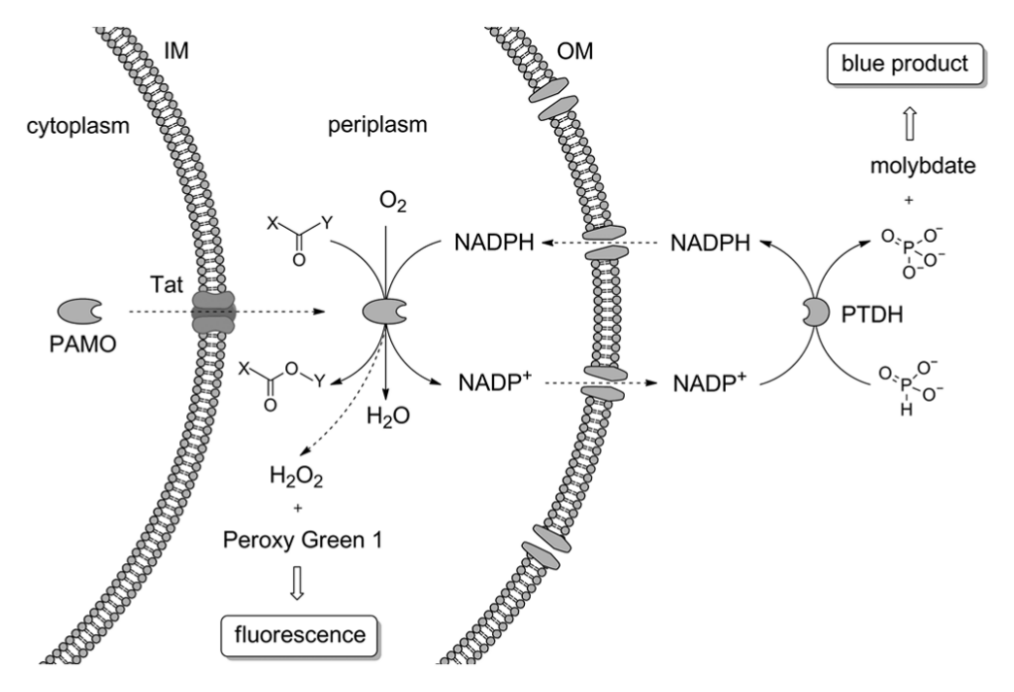
Schematic representation of the principle of the developed screening method. Phenylacetone monooxygenase (PAMO) is translocated to the periplasm by the Tat transporter. In the periplasm, PAMO performs the oxidation of a substrate at the expense of NADPH. NADPH and NADP+ can freely diffuse into and out of the periplasm. Phosphite dehydrogenase (PTDH) added to the reaction medium reduces NADP+ to NADPH while oxidizing phosphite to phosphate. The latter compound is measured in the reaction with molybdate by forming a blue product. Hydrogen peroxide, a product of the uncoupling reaction, is detected by fluorogenic Peroxy Green 1 (IM, inner membrane; OM, outer membrane).
Materials and Methods
Enzymes, Reagents, and Strains
Unless indicated, all chemicals were obtained from Sigma-Aldrich, Acros Organics, TCI Europe, Roche Diagnostics GmbH, and Merck. PfuTurbo was purchased from Strategene (La Jolla, CA). Restriction enzymes were obtained from New England Biolabs (Ipswich, MA). In-Fusion 2.0 Dry-Down PCR Cloning Kit (Clontech, Mountain View, CA) was used for DNA cloning. dNTPs were ordered at Clontech. Nucleotide primers were purchased at Sigma. NADPH was purchased at Oriental Yeast Co. Ltd. (Tokyo, Japan). Sodium phosphite was obtained from Riedel de Haën (Seelze, Germany). Peroxy Green 1 was kindly provided by P. Wiegerinck (Organon, The Netherlands). E. coli TOP10 cells were obtained from Invitrogen. DNA sequencing was performed at GATC Biotech (Konstanz, Germany). A modified low-phosphate AP5 medium 19 was used to grow E. coli cells expressing Tat-PAMO. The AP5 medium contained NaCl (60 mM), KCl (10 mM), NH4Cl (20 mM), MgSO4 (1.6 mM), triethanolamine (150 mM, pH 7.4), glycerol (0.15% v/v), and N-Z-amine A from bovine milk (0.3% w/v, Fluka). As compared with the original recipe, glucose was replaced by glycerol, and no phosphate source was added. Approximately 0.3 mM phosphate present in the medium comes from casein hydrolysate N-Z-amine A. All reagents used in the screening were prepared using filtered water. Molybdate reagent was prepared by dissolving zinc acetate (100 mM, Fluka) and ammonium molybdate ((NH4)6Mo7O24•4H2O, 10 mM, Sigma) and adjusting pH to 5.0. The solution was stored at room temperature in a plastic bottle and was stable for at least 6 mo. Ascorbic acid solution was prepared by dissolving L-(+)-ascorbic acid (10% w/v, Sigma-Aldrich) and adjusting pH to 5.0. It was aliquoted and stored at −20 °C. Stock solutions of phenylacetone (1 M), benzyl phenyl sulfide (0.125 M), and Peroxy Green 1 (25 mM) were prepared by dissolving the respective compounds in 1,4-dioxane.
Subcloning, Expression, and Purification of PTDH
A codon-optimized thermostable variant of the ptxD gene was amplified from the pCRE2 vector 20 and subcloned into the same vector using NdeI/HindIII sites. This yielded the pPTDH plasmid in which the ptxD gene is fused to an N-terminal hexahistidine tag. E. coli TOP10 cells were transformed with this plasmid and grown in LB supplemented with ampicillin (50 µg mL−1, LBAmp). The next day, the cells were diluted (1:100) in Terrific Broth supplemented with ampicillin (50 µg mL−1) and L-arabinose (0.02% w/v) and incubated at 30 °C for 16 h. The protein was purified using Ni Sepharose High Performance (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Purified PTDH was flash-frozen with the addition of glycerol (10% v/v) and stored at −80 °C. The concentration of PTDH was determined using two assays: Waddell’s method 21 and the estimated extinction coefficient at 280 nm: ϵ280 nm = 26.5 mM−1 cm−1 (http://web.expasy.org/protparam).
Export of PAMO to the Periplasm
The pamO gene was cloned into the previously described pBAD-Tat-AldO plasmid 22 via EcoRI and HindIII sites, replacing the aldO sequence in the vector. In this way, the pamO gene was fused to an N-terminal signal sequence from E. coli TorA protein and a C-terminal Myc epitope/histidine-tag. E. coli TOP10 cells were transformed with the Tat-PAMO plasmid. To test whether PAMO was present in the periplasm, the cells harboring Tat-PAMO plasmid were grown in LBAmp supplemented with L-arabinose (0.0002-0.005% w/v) at 17 °C or 24 °C for 48 h and at 37 °C for 16 h, and subsequently fractionated with the help of PeriPreps PeriPlasting kit (Epicentre Biotechnologies, Madison, WI). Specific activity of PAMO in spheroplast and periplasmic fractions was measured using the NADPH depletion activity assay, whereas the Bradford assay (Bio-Rad, Hercules, CA) was employed to assess the protein content in the samples. In the NADPH depletion assay, the reaction mix contained phenylacetone (1 mM), NADPH (100 µM), and DMSO (1% v/v). The reaction was followed by measuring the absorbance at 340 nm (ϵ NADPH, 340 nm = 6.22 mM−1 cm−1).
Expression of Tat-PAMO in 96-Well Plates
All optical measurements (optical density, OD600; absorbance; fluorescence) were conducted on a SynergyMx microtiter plate reader (BioTek Instruments, Inc., Winooski, VT) using appropriate plates purchased from Greiner Bio-One (Kremsmünster, Austria). Cells were routinely grown in 2 mL 96-square well plates (Waters, Milford, MA) covered with air-permeable AeraSeal Sealing Films (Excel Scientific, Victorville, CA). The plates were incubated in a Titramax 1000 shaker (Heidolph, Schwabach, Germany) at 1050 rpm. Precultures were inoculated from master plates using a cryo-replicator (Enzyscreen, Haarlem, The Netherlands) into a 96-well plate containing AP5Amp medium (1 mL) in each well. The plate was incubated at 30 °C for ~22 h. Afterwards, 60 µL of each preculture was diluted in fresh AP5Amp (800 µL) prewarmed to 37 °C and incubated at 37 °C for 110 to 120 min until the cultures reached the OD600 of 0.4 to 0.5. Then, L-arabinose (40 µL of 4% w/v solution) was added to the cultures along with FAD (9 µL of 1 mM solution). The plate was incubated at 37 °C for 16 h.
Screening Procedure
After overnight expression of Tat-PAMO, cells were pelleted by centrifugation for 20 min at 2250 × g at 4 °C and resuspended in reaction mixture (250 µL), which contained NADPH (20 µm), sodium phosphite (20 mM), 1,4-dioxane (1% v/v), sodium cyanide (20 mM), Peroxy Green 1 (250 µM), PTDH (5 µM), FAD (10 µM), Tris-HCl (50 mM, pH 9.0), and glycerol (10 mM). Afterwards, 10 µL of benzyl phenyl sulfide (0.125 M) was dispensed into each well. In the case of activity assays with phenylacetone, the reaction mixture contained the substrate (10 mM), NADPH (50 µM), sodium cyanide (20 mM), Peroxy Green 1 (250 µM), PTDH (5 µM), FAD (10 µM), Tris-HCl (50 mM, pH 7.5), and glycerol (10 mM). Reactions with phenylacetone were conducted for 1.5 h at 37 °C, 1050 rpm. In the case of benzyl phenyl sulfide, the reaction time was 1.5 or 3 h. Subsequently, the plate was centrifuged for 5 min at 2250 × g at 4 °C. A sample of the supernatent (100μL) was used to measure the fluorescence. Excitation was at 460 nm, and emission was recorded at 510 nm, while the sensitivity was set to 45. Mutants presenting at least 80% of the fluorescence emitted by the Q152F/L153G/M446G mutant were regarded as the uncoupling mutants and not considered for further analysis. For the quantification of phosphate, the supernatant (20 µL) was mixed with the molybdate reagent (200 µL), followed by the addition of ascorbic acid (50 µL). After 30 min incubation at 30 °C and 900 rpm, absorbance was read at 850, 700, and 600 nm. Phosphate standards containing potassium dihydroxy phosphate (from 0.2 to 10 mM) were used to prepare the calibration curve for the molybdate assay. The Z′ factor was used as a measure of the quality of the assay during the optimization, and it was calculated according to the formula introduced by Zhang et al. 23
Results
Expression of PAMO in the Periplasm
We sought to develop a generic screening procedure for NAD(P)-dependent enzymes based on whole cells, using PAMO as a model BVMO. To be successful, this system should offer unrestricted access of the enzyme toward the substrate tested. Unfortunately, many (potential) substrates are unable to cross the cytoplasmic membrane and therefore cannot react with the target enzyme expressed in the cytoplasm. Several strategies have been presented for E. coli to increase substrate accessibility. Secretion of the target enzyme out of the cytoplasm to the periplasm is one of the typical approaches. The Tat pathway is the only one among bacterial translocation systems that is capable of transporting fully folded and often cofactor-containing proteins to the periplasm and is therefore of considerable biotechnological importance. 24 Considering that PAMO requires non–covalently-bound FAD cofactor for its activity, we decided to study whether the Tat pathway could be employed for functionally transporting PAMO to the periplasm. To this end, PAMO was N-terminally fused to the Tat-dependent signal sequence of the endogenous E. coli protein TorA, 25 resulting in Tat-PAMO. Subsequently, we determined the best conditions for periplasmic expression of Tat-PAMO. For this, Tat-PAMO was expressed in E. coli under control of an arabinose-inducible promoter using increasing amounts of arabinose at 17 °C, 24 °C, or 37 °C. These cells were harvested and fractionated into spheroplast and periplasmic fraction. Of note, the spheroplast fraction contains the cytoplasmic and total membrane fraction. Next, we analyzed the specific activities of these fractions using phenylacetone as a substrate ( Fig. 2 ). At 17 °C and 24 °C, the highest activity toward phenylacetone was clearly observed in the spheroplast fraction relative to the periplasmic fraction regardless of the arabinose concentration. However, at 37 °C, the differences in the observed activities between the different fractions became less pronounced. In particular, a slightly higher specific activity for phenylacetone was observed in the periplasmic fraction derived from cells induced for Tat-PAMO expression with 0.0002% arabinose relative to the spheroplast fraction prepared from the same cells. This indicates that PAMO was indeed for a significant part translocated to the periplasm.
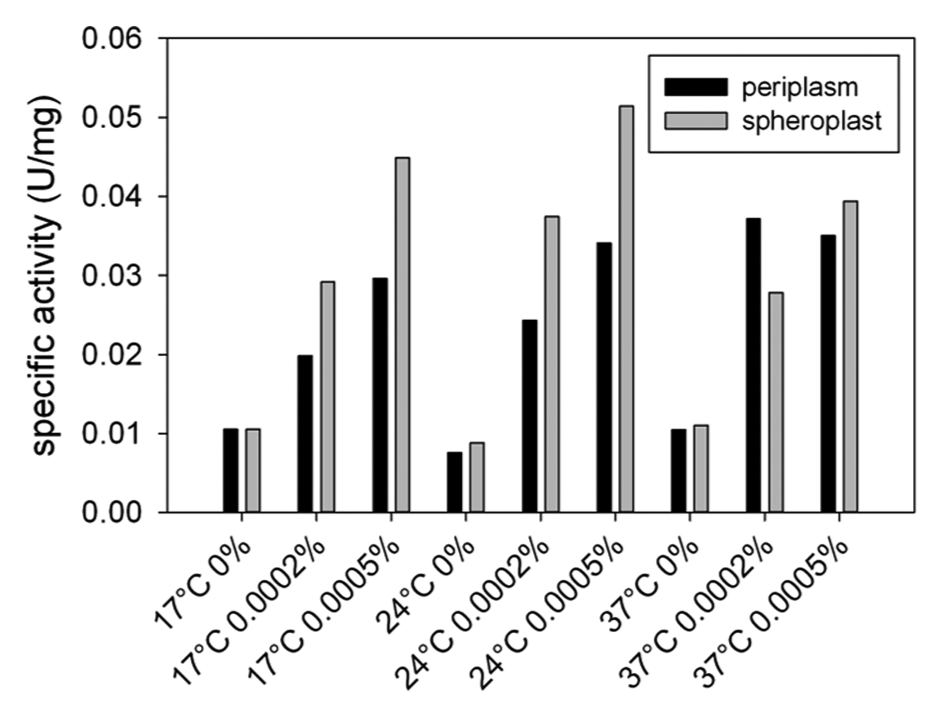
Specific activity of periplasmic and spheroplast fractions of phenylacetone monooxygenase (PAMO) toward phenylacetone. Escherichia coli cells expressing Tat-PAMO were fractionated into periplasm and spheroplast fractions. The concentrations of arabinose and temperatures used for the expression of Tat-PAMO are detailed.
Although our data show that attachment of the TorA signal sequence does not inactivate PAMO, it cannot be excluded that this compromises the enzyme to some extent. Therefore, we decided to analyze the properties of PAMO isolated from a periplasmic preparation in more detail. First, we found that the enzyme obtained from a periplasmic preparation runs at the same position in sodium dodecyl sulfate polyacrylamide gel electrophoresis as native PAMO expressed in the cytoplasm, indicating that the TorA signal sequence is proteolytically removed during periplasmic export as shown for other Tat-dependently exported proteins. 24 Second, the kcat and KM values for phenylacetone of PAMO expressed in the periplasm did not differ from the kinetic parameters of WT PAMO. Last, the apparent melting temperature of the mature-sized periplasmic form measured by the ThermoFAD method 26 was the same as of WT PAMO (60 °C). This confirms that the function of PAMO is not affected by fusing it to the TorA signal sequence.
Culture Conditions
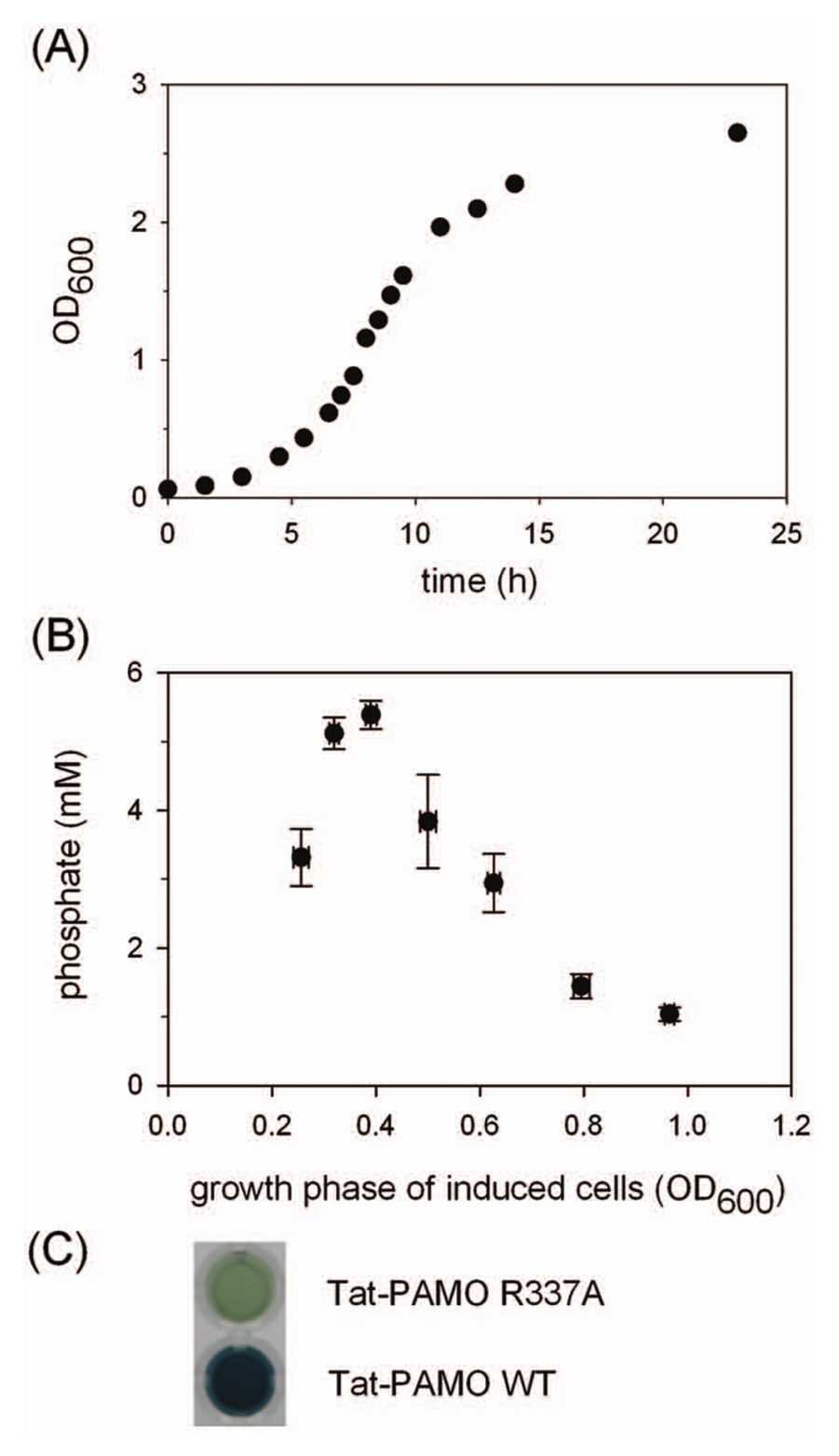
Subsequent experiments were aimed at optimizing the conditions for the periplasmic export of PAMO. To this end, we developed an activity-based assay in 96-well plate format revolving around phosphate detection. After evaluating different growth media for our screening method, we obtained the best results with a modified version of AP5, a defined, low-phosphate medium. 19 The phosphate content in this medium was found to be only ~0.3 mM phosphate compared to, for example, ~4 mM in LB medium. The low amount of phosphate in AP5 ensures low background signals without the need of washing cells before the activity assay. Importantly, the growth of E. coli is not compromised by the limited amount of phosphate in liquid AP5 medium, as evidenced by the optical density of 2.5 to 3 when cells are grown to saturation in AP5 ( Fig. 3A ).
(
To effectively control the growth of cells in individual wells, the optimized protocol involved growing precultures inoculated with cells from a glycerol stock or fresh transformants. The precultures were then diluted in fresh AP5 medium and grown for a given time before arabinose was added to induce the production of Tat-PAMO. Previously, we assessed several critical factors that control the reproducible expression of PAMO, and we observed that the cellular growth stage at which induction is initiated is of crucial importance for good overexpression. 27 Therefore, we also determined the best induction time for the expression of Tat-PAMO. Furthermore, in the case of periplasmic expression, the amount of the protein in the periplasm depends not only on the overall expression level but also on the efficiency of the export to the periplasm. In other words, the highest total expression level may not correspond to the highest periplasmic expression. Therefore, we evaluated the induction protocol by measuring the whole-cell activity of Tat-PAMO cells with the phosphate assay. The highest activity was observed when cells were induced during the early exponential phase ( Fig. 3B ). Because we observed a clear peak in activity depending on the growth phase, care had to be taken to induce the cells always in the same growth phase. We achieved this by using high-volume inocula, medium prewarmed to 37 °C, and precise culturing times.
We analyzed whether external addition of FAD would improve the activity of the enzyme because it cannot be excluded that the enzyme partially loses its cofactor when it is placed in the environment lacking any free FAD such as the periplasm. Therefore, we tested several conditions in which 10 or 100 µM FAD was added to the culture medium during the expression and/or to the reaction medium during the biotransformation. Surprisingly, we observed that the activity of Tat-PAMO can be doubled by adding 10 µM FAD during both the expression and the biotransformation. This indeed suggests that PAMO partially loses FAD in the periplasm.
Coenzyme Regeneration System
We have previously reported on an elegant system for coenzyme regeneration suitable for BVMOs by fusing them to PTDH.16,20 We sought to use the same coenzyme regeneration system for the design of our screening method. Under normal conditions, namely, no phosphate limitation, phosphite does not enter the cytoplasm of E. coli because pathways for assimilation of alternative phosphorus forms are repressed.28,29 However, under these conditions, phosphite is most likely able to cross the outer membrane and enter the periplasmic space. Export of PTDH to the periplasm is therefore required to enable efficient cofactor recycling in this cellular compartment. However, appending PTDH to the N-terminus of Tat-PAMO impaired Tat-mediated transport of this fusion protein (data not shown). Therefore, we decided to use the established periplasmic expression of PAMO in combination with externally added PTDH because NADP(H) can diffuse into and out of the periplasm through the outer membrane pores. To facilitate overexpression and purification, PTDH was subcloned into the pBAD vector with an N-terminal histidine-tag. It could be purified on Ni2+-resin, yielding up to 600 mg of protein per 1 L medium. In this way, the export of PAMO was not compromised by addition of the fusion partner. Furthermore, by adding isolated PTDH to the reactions, the concentration of the enzyme can be manipulated.
Phosphate Detection in Molybdate Assay
Methods commonly used for quantification of inorganic phosphate employ ammonium molybdate that reacts with phosphate to form a chromophore. These methods are typically modifications of a method published by Fiske and Subbarow, 30 and they require the use of strong acids, which is not compatible with biological samples. Therefore, we adapted a method of Saheki et al., 17 which was developed for assaying biological samples and is performed in slightly acidic conditions. In this method, the phosphomolybdate complex is reduced with ascorbic acid in the presence of Zn2+ ions, which leads to the formation of a blue product with an absorbance maximum at about 850 nm. As compared with the protocol of Saheki et al., 17 we decreased the ammonium molybdate concentration from 15 to 10 mM and increased the time of incubation at 30 °C from 15 to 30 min. The color formed is stable for at least 1 h. We down-scaled the procedure to make it compatible with 96-well plate format. A linear dependence between the phosphate concentration and the absorbance was observed at up to 4 mM phosphate. For phosphate concentrations higher than 4 mM, the absorbance exceeded the detection limit. By shifting the detection wavelength to 600 nm, we extended the working range of the assay up to 10 mM phosphate. If required, active and inactive clones can be distinguished by direct visual inspection, as presented in Figure 3C .
Control of the Uncoupling
It has been observed for BVMOs that in the absence of a suitable substrate, the peroxyflavin intermediate can decay, forming hydrogen peroxide. 18 In this way, a BVMO can act as an NADPH oxidase. This process, referred to as uncoupling, is unfavorable in our screening for two reasons. First, mutants with a high uncoupling rate may give high signals in the screening while they perform the desired conversion at a low rate or are completely inactive in the Baeyer-Villiger reaction. Thus, uncoupling may result in the isolation of false-positive clones in our NADPH-consumption–based screening system. Second, enzymes presenting increased uncoupling rates, even when they are still able to catalyze the reaction, work less efficiently because of the elevated coenzyme requirements and often reduced catalytic rates. Meanwhile, mutations introduced in the active site of the enzyme in order to alter its catalytic properties may affect the stability of the peroxyflavin, thereby increasing the uncoupling rate. These reasons prompted us to include a control for hydrogen peroxide detection in order to increase the fidelity of our method. To this end, we decided to test Peroxy Green 1. It has been established that this dye is a highly specific probe for hydrogen peroxide. 31 In the presence of hydrogen peroxide, the boronate moiety is cleaved off, leading to the formation of fluorescent 2-methyl-4-O-methyl Tokyo Green.
For establishing the conditions of the fluorescence measurements, we selected the previously characterized Q152F PAMO mutant 32 and the triple Q152F/L153G/M446G PAMO mutant. 33 Both mutants exhibit high uncoupling rates (0.5–0.6 s−1) relative to WT PAMO (0.02 s−1). 18 These mutations were subsequently introduced in Tat-PAMO. We found out that the cells producing these Tat-PAMO variants show Peroxy Green 1–based fluorescence values higher than WT Tat-PAMO only if catalase activity is inhibited by adding sodium cyanide to the reaction medium. Conceivably, the increased amounts of hydrogen peroxide produced in these cells induce the expression of catalase activity. Nevertheless, in the presence of Peroxy Green 1 and sodium cyanide, a clear increase in the fluorescence emitted by the uncoupling mutants as compared with WT PAMO was observed. Furthermore, addition of Peroxy Green 1 or sodium cyanide did not interfere with the phosphate detection. These findings show that Peroxy Green 1 can be incorporated in the phosphate screening method for efficient hydrogen peroxide detection, which allows elimination of variants with high uncoupling rates.
Evaluation of the Screening Method
After having established the best conditions for the expression of Tat-PAMO as well as the conditions for our screening method, we tested whether this optimized protocol could be used reproducibly to distinguish cells expressing the previously described PAMO mutant R337A from cells expressing WT PAMO, using phenylacetone as substrate. The R337A mutant is inactive in Baeyer-Villiger reaction and consumes NADPH at a very low rate (0.001 s−1). 18 To this end, we inoculated 32 wells with single colonies of E. coli cells transformed with a Tat-PAMO expression plasmid and 32 wells with cells harboring a Tat-PAMO-R337A expression plasmid. Each of these precultures was subsequently used to inoculate an expression culture, which was then assayed for BVMO activity, as described in the Materials and Methods section. The amounts of phosphate produced by each culture are shown in Figure 4A . In the control reaction of WT PAMO, the mean signal of 5.0 mM was obtained while the background signal given by the inactive mutant R337A was only 0.54 mM ( Table 1 ). This background signal can be caused by NADPH-consuming enzymes released from lysed cells. Relative standard deviations calculated from these data did not exceed 12%, which is a satisfactory value for a whole-cell–based system. Furthermore, these data correspond to a Z′ factor value of 0.6, which indicates very good separation of the signals and confirms the reliability of our screening method. 23
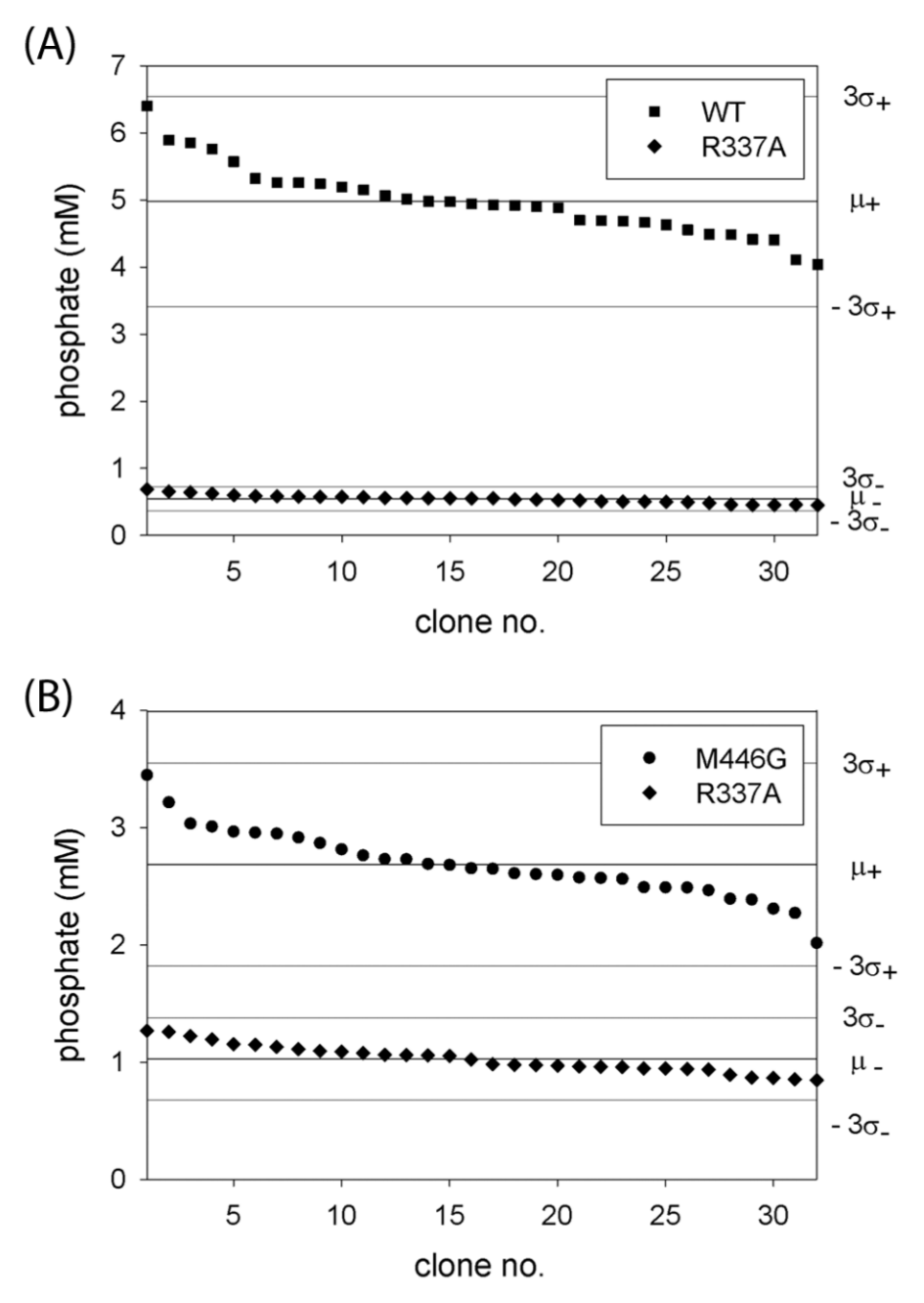
Phosphate signals obtained in independent replicates of the reaction of WT phenylacetone monooxygenase and the R337A mutant with phenylacetone (
Reproducibility of the Screening Method. a
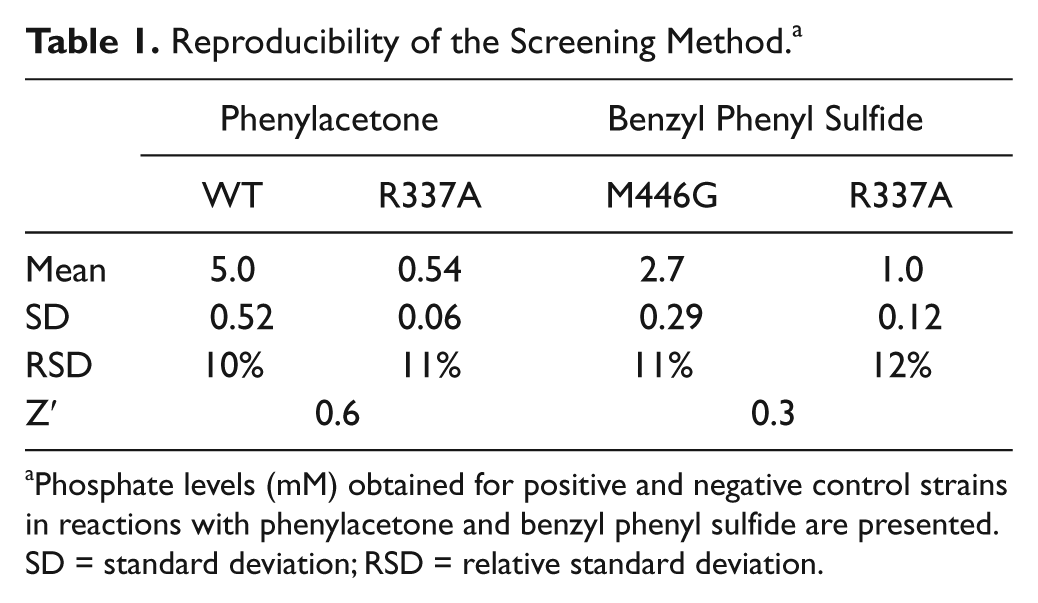
Phosphate levels (mM) obtained for positive and negative control strains in reactions with phenylacetone and benzyl phenyl sulfide are presented. SD = standard deviation; RSD = relative standard deviation.
Encouraged by these results, we used the same experimental set up to test whether our method could be used to distinguish cells expressing the PAMO mutant M446G from cells expressing the R337A variant using benzyl phenyl sulfide as substrate. This prochiral bulky compound is not converted by WT PAMO, whereas the M446G mutant has been shown to oxidize it to the corresponding sulfoxide. 32 To compensate for the low conversion rate, the reaction was performed at pH 9.0, at which the activity of PAMO is increased relative to pH 7.5. Concentration of 1,4-dioxane in the reaction mixture was increased to 6% v/v to facilitate the solubilization of the substrate. This amount of organic solvent did not inactivate either PAMO or PTDH. The reaction was conducted for 90 min. As shown in Figure 4B , the signals given by the M446G mutant can be clearly distinguished from the background signals. Not surprisingly, the separation band is smaller than in our previous control reaction of the wild-type enzyme with phenylacetone, but this reaction reflects better the situation of screening for activity on a challenging substrate. The relative standard deviations were at the level of 11% to 12%, whereas the calculated Z′ factor value was 0.3 ( Table 1 ), indicating that the method still allows accurate screening.
To verify whether the measured phosphate levels correspond to the actual product formation, we employed GC for the quantification of the Baeyer-Villiger product in parallel with phosphate measurements. Because of the adsorption of both phenylacetone and benzyl acetate onto plastic, we used glass vials mimicking the conditions in wells of a 96-well plate. A total of 5.0 ± 0.8 mM of benzyl acetate was produced by Tat-PAMO cells in the presence of the coenzyme regeneration system as determined by GC analysis. Control experiments in the absence of the regeneration system showed that only traces of benzyl acetate (<0.1 mM) were detected. This indicates that Baeyer-Villiger activity of Tat-PAMO cells is related to the periplasmic fraction of the enzyme, which is dependent on the external coenzyme regeneration. When the same samples were analyzed using the phosphate detection assay, 5.4 ± 1.2 mM of phosphate was measured. Only 0.6 ± 0.2 mM of phosphate is produced by the R337A variant in this reaction, whereas no oxidized product was detected by GC analysis. This indicates that coenzyme recycling drives the production of about 4.8 mM phosphate by a BVMO, demonstrating the effectiveness of our coenzyme regeneration system. Thus, the obtained values of phosphate and benzyl acetate production under the assay conditions are within 10% of each other. These results confirm the applicability of phosphate as a reporter of BVMO activity in our system.
Screening Libraries of PAMO
The ability of our screening method to distinguish active PAMO variants from inactive ones demonstrates proof of principle. Therefore, the screening method was applied in screening of two site-saturation libraries of Tat-PAMO. In these libraries, the positions I67 or M446 were randomized, and for both libraries, 91 randomly picked clones were assayed. Both libraries were tested for activity with benzyl phenyl sulfide, which is poorly accepted by WT PAMO. 32 In the screening of the M446X library, a reaction time of 90 min was applied, and clones producing at least 1.4 mM phosphate were considered as active. To minimize false-positive hits, we excluded clones displaying the fluorescence of at least 80% of the Q152F/L153G/M446G mutant. Using these threshold values, eight active clones were isolated from the M446X library. Of these hits, the previously characterized M446G variant was found seven times, while an M446A mutant was also found. To analyze the properties of the newly found M446A mutant in more detail, both enzymes were isolated and their reactivity toward benzyl phenyl sulfide was tested. The M446A mutant proved to convert this substrate with higher efficiency than the M446G variant (70% conversion in the case of M446A and 59% conversion in the case of M446G) and similar excellent enantioselectivity, leading to the (R)-sulfoxide as the main product (in both cases more than 98% ee). The I67X library was screened using less stringent threshold values, because initial experiments showed overall lower activity among mutants in this library. Therefore, a reaction time of 3 h was applied, and clones producing at least 0.8 mM phosphate were further analyzed. Under these conditions, 11 clones were isolated, and 10 of these were found to be the I67S variant, whereas I67T was obtained once. Both mutants were purified, and their reactivity toward benzyl phenyl sulfide was studied. Although the ability of I67T to convert benzyl phenyl sulfide has been reported before, 32 the I67S mutant also produced the sulfoxide with high enantiomeric excess (98% ee for the (S)-enantiomer).
We also picked several clones displaying the highest fluorescence (NADPH oxidase activity) in each library. It turned out that all four uncoupling clones selected from the I67X library were I67P mutants. Three uncoupling mutants from the M446X library contained an M446W mutation. These mutations, that is, the introduction of a bulky residue at position 446 or a proline instead of an isoleucine at position 67, are indeed expected to perturb the active site architecture, resulting in uncoupling mutants, which thereby further confirms the accuracy and the reliability of our screening method.
Discussion
Directed evolution experiments often lead to the conclusion that the screening conditions should reflect as much as possible the real conditions under which the reaction is to be performed. Use of surrogate or labeled substrates may result in identification of mutants less active on the target compound or not active at all. Furthermore, activity assays are often based on the particular property of a substrate or a product and thus need to be established for each compound. This underscores the need for a generic screening procedure applicable to a given class of enzymes and that is compatible with unmodified substrates.
BVMOs can perform different types of oxidations: Baeyer-Villiger oxidation, sulfoxidation, epoxidation, and oxidation of heteroatoms such as nitrogen, boron, and selenium.1,2 A truly generic method for this class of enzymes would enable screening for all these different activities. Therefore, a generic method can be related to oxygen or NADPH consumption by BVMOs during catalysis. However, accurate measurements of oxygen depletion would be difficult to perform in a high-throughput manner because oxygen diffuses easily, and a tight closure of the reaction system would be required. Moreover, oxygen is consumed by cellular metabolism, which would interfere with a whole-cell activity screening method based on oxygen consumption. On the other hand, following NADPH depletion is a routinely used activity assay for BVMOs. To apply this method for screening libraries of mutants, cell extracts need to be prepared first because there are no transporters for NADPH in the cell membrane. Lysing cells is a time- and labor-consuming step. Moreover, cellular NADPH-dependent enzymes are released, which interferes with the measurement, and high expression levels or high activities of the targeted enzyme are necessary to reliably detect its activity in the cell extract. Lastly, the absorbance spectrum of NADPH overlaps with spectra of many potential substrates, which would hamper accurate determination of NADPH.
Here, we have addressed the above-mentioned issues and have developed a generic, whole-cell–based procedure suitable for screening BVMOs in 96-well plate format. Our method revolves around periplasmic expression of the target enzyme in E. coli because periplasmic location offers unrestricted access of the enzyme toward the substrate tested, as has been shown in previous studies. 34 In addition, directing the enzyme outside the cytoplasm helps to separate the activity of interest from cytoplasmic enzymes, which can interfere with the activity assay. Based on the cofactor requirements of PAMO, we decided to use the Tat pathway for periplasmic transport. Our data indeed show that PAMO is transported to the periplasm in a functional form. Importantly, PAMO is the first flavoprotein monooxygenase expressed functionally in the periplasm.
Moreover, periplasmic expression of PAMO enables efficient coenzyme recycling by the established PTDH-based regeneration system,16,20 using externally added PTDH. We were able to detect and quantify the phosphate released by this system during NADPH regeneration in a colorimetric reaction with molybdate, 17 allowing the use of the released phosphate as a reporter of BVMO activity. In this way, we could accurately measure BVMO activity using whole cells. The method allows endpoint measurements, and the detected phosphate amount corresponds to the amount of the oxidation product. Although it is possible to spectrophotometrically observe NADPH consumption by E. coli cells expressing PAMO in the periplasm, the accuracy is affected by light-scattering effects of cells. Here, by using indirect measurements of NADPH depletion, we can move the detection to the visible range and use high cell densities. Employing the regeneration system further increases the sensitivity of the method because the amount of phosphate can build up during multiple turnovers. This principle could also be applied to cell extracts, but in this case, the background would be much higher because of cellular NADPH-consuming enzymes.
BVMOs are known to produce H2O2 in the absence of a suitable substrate while consuming NADPH. This form of nonproductive catalysis is undesirable in our screening method because it increases the number of false-positive hits. Although in WT PAMO uncoupling occurs at a low rate, 18 this reaction can be promoted by mutations introduced in the active site during creating libraries. To increase the accuracy of our screening method, we included an additional control reaction to monitor the formation of H2O2. To this end, we successfully used Peroxy Green 1, a fluorescent dye highly specific for H2O2. 31 It is worth noting that our work represents the first example of using Peroxy Green 1 in mutant library screening.
We showed that the procedure enables distinguishing between active PAMO variants and inactive variants and that the amount of phosphate produced during biotransformation is in agreement with the amount of the oxidation product. This clearly demonstrated proof of principle, thereby validating our method and confirming its robustness. Subsequently, two site-saturation libraries of Tat-PAMO, randomized at positions I67 or M446, were successfully screened using our protocol, resulting in the identification of two novel PAMO variants with interesting enantioselectivities. Notably, screening of other libraries using this procedure is currently ongoing in our laboratory.
Our newly developed method offers the advantage of screening for the activity toward unmodified substrates. In fact, the same protocol can be employed for any potential substrate. In this report, we have used the method in screening for activity on phenylacetone and benzyl phenyl sulfide. These substrates undergo different reaction types, namely, the Baeyer-Villiger oxidation and sulfoxidation. Because the enzyme is expressed in the periplasm, which is a weakly controlled cellular compartment, it is possible to screen for activity or stability in different reaction media (for example, at pH different than neutral or in the presence of solvents). This points to the flexibility of the protocol. Moreover, the applications of the new method are much broader than just screening for BVMO activity. In principle, the method can be applied in the screening of any NADPH-consuming enzyme. Moreover, because PTDH accepts both NADPH and NADH, this method can be used to screen for NADH-dependent enzymes as well, or it could be employed to evolve enzymes with altered coenzyme specificity. Even though the method is based on the general ability of enzymes to oxidize NADPH, it allows immediate elimination of variants catalyzing unproductive cycles, generating hydrogen peroxide. The established screening protocol is suitable for 96-well plates, which obviously limits the throughput to several hundred clones per day. We are currently exploring options to translate the reported principle of screening to a higher throughput format, for example, by growing and screening colonies on agar plates.
Footnotes
Acknowledgements
We thank Daniel Torres Pazmiño and Remko Winter for helpful discussions.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. Wouter A. Duetz has a financial interest in Enzyscreen BV.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research leading to these results has received funding from the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement n°212281.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.