
Abstract
Ribonuclease H2 (RNase H2) is a nuclease that specifically hydrolyzes RNA residues in RNA-DNA hybrids. Mutations in the RNase H2 enzyme complex have been identified in the genetic disorder Aicardi-Goutières syndrome (AGS), which has similarities to the autoimmune disease systemic lupus eryrthrematosis (SLE). The RNase H2 enzyme has also been recently implicated as a key genome surveillance enzyme. Therefore, small-molecule modulators of RNase H2 activity may have utility in therapeutics and as tools to investigate the cellular functions of RNase H2. A fluorescent quench assay, measuring cleavage of an RNA-DNA duplex substrate by recombinant RNase H2, was developed into a high-throughput format and used to screen a 48 560 compound library. A hit validation strategy was subsequently employed, leading to the identification of two novel inhibitor compounds with in vitro nanomolar range inhibition of RNase H2 activity and >100-fold selectivity compared with RNase H type 1. These compounds are the first small-molecule inhibitors of RNase H2 to be reported. They and their derivatives should provide the basis for the development of tool compounds investigating the cellular functions of the RNase H2 enzyme, and, potentially, for pharmacological manipulation of nucleic acid–mediated immune responses.
Introduction
There are two mammalian ribonuclease (RNase) H nucleases (RNase H1 and RNase H2), which can hydrolyze the RNA strand of RNA/DNA hybrids. RNase H2 is the major source of ribonuclease H activity in the cell, 1 and so is important for the hydrolysis of physiological RNA:DNA hybrids that form within the cell. Substrates may include the RNA primers that initiate Okazaki fragment synthesis during lagging strand DNA replication. 2 In addition, RNase H2 can specifically cleave the phosphodiester bond 5′ of a single ribonucleotide embedded in a DNA duplex3,4 and is essential for the removal of such ribonucleotides misincorporated in genomic DNA during replication.5–8 RNase H2 is an enzyme complex consisting of three subunits, all of which are required for enzyme function.4,9,10 Mutations that impair human RNase H2 function have been identified in the autoimmune neurological disorder Aicardi-Goutières syndrome (AGS), which causes a severe progressive neurological disability with onset in infancy and has clinical similarities to congenital viral infection. 9 Reduced RNase H2 function is thought to trigger inflammation through the abnormal accumulation of immunogenic intracellular nucleic acids, and it has been proposed that those nucleic acids are recognized by pattern recognition receptors, whose function is to detect viral infections.11,12 The immunological features of AGS are similar to the autoimmune disease systemic lupus erythematosis (SLE), in which nucleic acid–mediated inflammation is also implicated. 13 Notably, mutations in TREX1, one of the other nucleases causing AGS, are found in a proportion of SLE patients. 14
RNase H2 has also been suggested as a putative anticancer drug target, based on its overexpression in cancer cell lines and findings of a genome-wide siRNA screen. 15 siRNA depletion of RNase H2 resulted in inhibition of anchorage-independent growth but not proliferation in vitro. Significantly, it has recently been established that RNase H2 is required for maintaining genome stability in mammals,7,8 where it acts as a genome surveillance enzyme to remove ribonucleotides misincorporated by replicative polymerases. Such ribonucleotides were also found to be the most common endogenous nucleotide base lesions in replicating cells. 7
Therefore, small molecules that modulate the activity of RNase H2 could be useful research tools, relevant both to understanding autoimmune disease pathogenesis and the maintenance of genome integrity. In addition, further insights into the cellular function of RNase H2 could aid in the discovery of therapeutic agents for AGS and related autoimmune diseases. Although activators of RNase H2 may be therapeutically beneficial to normalize enzyme function in AGS, the utility of inhibitors is likely to be limited given the enzyme’s role in maintaining genome stability. Given that the HIV-1 reverse transcriptase RNase H has successfully been used as a target in high-throughput screening (HTS) to identify new leads for new antiviral agents,16–18 we performed a similar screen for the human RNase H2 enzyme.
Here we describe the development of an RNase H2 enzyme assay for HTS of small-molecule libraries to identify compounds that inhibit or activate RNase H2 enzyme activity. A hit validation strategy was also established, employing enzyme and cellular assays and profiling hit compound properties. Using these assays, we identified several compounds that inhibited RNase H2 activity to nanomolar compound IC50 concentrations and were selective to RNase H2 over other nucleases.
Materials and Methods
Materials
Recombinant RNase H2 was prepared as described previously,
10
and 1 U was defined as the amount of enzyme that cleaves 20% of the 25 pmol substrate present in a 100 µL reaction in 90 min at 20 °C under standard fluorometric assay conditions. Km and Kcat for RNase H2 with the H2 substrate were 28 nM and 24 min−1, respectively, for the H2 substrate.
10
Recombinant type 1 RNase H (RNase HI) was purchased from New England Biolabs (Ipswich, MA). RNase A and DNase I were purchased from Roche (Basel, Switzerland). For initial assay validation, the NINDS collection of 1040 pharmacologically active compounds was used. Subsequently, the following compound collections were screened: a diversity set of ~40 000 compounds and 8500 kinase templates compiled by MRC Technology from commercial supplier collections and a natural product collection of ~3500 compounds sourced from Phytoquest and Analyticon. Hit compounds were sourced from original suppliers (
HeLa cell lysate
HeLa cells were grown to confluency using standard techniques, trypsinized, and spun at 232g for 5 min. Pellets were washed in phosphate-buffered saline and spun at 232g for 5 min, resuspended in whole cell extract buffer (50 mM Tris pH 8, 280 mM NaCl, 0.5% NP40, 0.2 mM EDTA, 0.2 mM EGTA, 10% glycerol, 0.1 mM Na Vanadate) and incubated on ice for 10 min. An equal volume of cytoplasmic buffer (20 mM Hepes pH 7.9, 10 mM KCl, 1 mM EDTA, 10% glycerol, 0.1 mM Na Vanadate) was added, incubated on ice for 10 min, and spun for 10 min at 13 793g. The lysate supernatant was removed and stored at −20 °C. Protein concentrations were determined using Bradford reagent (Bio-Rad, Hercules, CA).
Fluorometric substrates
A total of 10 µM of 3 ′fluorescein-labeled oligonucleotides (Eurogentec, Liege, Belgium) was annealed to 10 µM complementary 5′ dabcyl-labeled oligonucleotides in 60 mM KCl, 50 mM Tris-HCl pH 8, by denaturation at 95 °C for 5 min and gradual cooling to room temperature (20 °C): H2 substrate (GATCTGAGCCT-GGGaGCT upper strand, AGCTCCCAGGCTCAGATC lower strand; uppercase denotes DNA; lowercase, RNA), H1 substrate (gatctgagcctgggagct upper strand, DNA complementary lower strand), dsRNA substrate (gatctgagcctgggagct upper strand, RNA complementary lower strand), dsDNA substrate (GATCTGAGCCTGGGAGCT upper strand, DNA complementary lower strand).
RNase H2 Fluorometric Assays
Unless otherwise indicated, assays were performed in 100 µL RNase H2 assay buffer (60 mM KCl, 50 mM Tris-HCl pH 8, 10 mM MgCl2, 0.01% bovine serum albumin, 0.01% Triton X-100) with 250 nM substrate and 1 U of RNase H2 enzyme in 96-well black flat-bottom plates at room temperature (20 °C) for 90 min, with a final concentration of 20 mM EDTA being used to quench the reaction. Fluorescence intensity was detected for 100 ms using a Victor 2 1420 multilabel counter (Perkin Elmer, Boston, MA), using a 480 nm excitation filter and 535 nm emission filter.
10 mM aurintricarboxylic acid (Sigma, St. Louis, MO) and 400 U RNasin (Promega, Madison, WI) were used as controls for DNase I and RNase A assays, being known inhibitors of the respective enzymes.
HTS mode with 384 wells
The above assay was miniaturized to a final volume of 20 µL and automated using Biomek FX (Beckman Coulter, Fullerton, CA) for compound additions and Flexdrop (Perkin Elmer) for reaction initiation and stop. Compounds were screened at 10 µM (1% DMSO) and preincubated with 1 U (~2.3 fmol) RNase H2 for approximately 30 min prior to addition of H2 oligonucleotide substrate. The specific activity of recombinant RNase H2 used in the HTS screen was 1 000 000 U/mg. During this time, a preread at fixed gain settings was read to identify any autofluorescent compounds. The reaction was incubated for 90 min at room temperature and terminated by the addition of 10 µL 60 mM EDTA. Controls were run in columns 1, 2, 23, and 24 to enable detection of both activators and inhibitors and 320 single compound/well were run in columns 3 to 22. Plates were read using a PHERAstar plate reader (BMG-Labtech, Ortenberg, Germany). Data were normalized as a percentage of the DMSO control (1 U enzyme + 1% DMSO) – low control (substrate alone). A high control (2.5 U RNase H2) was used to calculate Z′ (relative to low control), and an additional quality control parameter was applied to pass plates, which was that more than 14/16 high controls must be greater than 3sd cutoff of DMSO-only control. Plates failing either Z′ (<0.5) or 3sd cutoff criteria were repeated.
Assay interference counterscreens
Hit compounds were added to either 250 nM H2 substrate or buffer alone, and fluorescence quench or activation was detected using the Victor 2 reader.
Compound stability assay
Each test compound was diluted to 100 µM in RNase H2 assay buffer at 24, 6, and 0 hours before assay was to be performed and maintained at room temperature. A final concentration of 10 µM of test compound was added to a total 100 µL assay volume containing 2.5 U recombinant RNase H2. This was preincubated at room temperature for 20 min, the reaction started with 250 nM H2 substrate, and incubated for 90 min at room temperature. Fifty microliters of 60 mM EDTA was used to quench the reaction, and plates were read using a Victor 2 reader.
Hit Confirmation Assays
Polyacrylamide gel electrophoresis assay
Two units of recombinant RNase H2 was incubated with 25 µM test compound for 30 min at room temperature in the dark. H2 substrate 625 nM (without a 5′-dabcyl on the reverse strand) and 10% glycerol were added at room temperature, after 10 min incubation samples were loaded onto an 8% novexpolyacrylamide gel (Life Technologies, Grand Island, NY) and run at 100 V for 1 h at 4 °C. Fluorescently labeled substrate and cleavage products were detected and quantified using an FLA-5100 phosphorimager (GE Healthcare).
Cell viability assay
A total of 1.25 × 104 HeLa cells per well were added to a 96 well µclear plate (Greiner, Kremsmunster, Austria) and grown overnight in DMEM (Life Sciences) with 10% fetal calf serum at 37 °C 5% CO2. A final concentration of 10 µM test compound was added to each well and incubated overnight at 37 °C, 5% CO2. Twenty microliters of Cell Titre Blue reagent (Promega) was then added to each well, the plate agitated for 10 s, and incubated for 1 h at 37 °C, 5% CO2, before being read on the Victor 2 at 560/590 nm.
Results and Discussion
Optimization of a Fluorescence Resonance Energy Transfer–Based Fluorometric RNase H2 Assay for HTS
A fluorescent-based enzyme activity assay has previously been established for RNase H2, 9 which was adapted from an assay for HIV-1 RT-RNase H. 16 The assay uses a duplex oligonucleotide substrate consisting of a 3′-fluorescein-labeled oligonucleotide annealed to a 5′-dabcyl-labeled DNA oligonucleotide, with the dabcyl group quenching the fluorescence from the adjacent fluorescein ( Fig. 1A , B ). RNase H2 hydrolysis of the substrate results in the release of the fluorescein-tagged oligonucleotide from the duplex and consequently enhanced fluorescent signal. In optimized conditions, this can be used as a highly sensitive kinetic assay of RNase H2 activity ( Fig. 1C ). To permit HTS, the kinetic assay was adapted to endpoint readout at 90 min by chelation of the Mg2+ cations required for catalysis with a saturating concentration of EDTA. To optimize the endpoint assay, the activity of recombinant RNase H2 was measured by testing different concentrations of recombinant RNase H2 protein complex against a set concentration of substrate at room temperature (20 °C) to ensure linear reaction and nonlimiting conditions of substrate turnover. The fluorometric assay was also similarly optimized to measure RNase H2 activity in crude HeLa cell lysates and to assay DNase I and RNase A against dsDNA and dsRNA substrates, respectively, for downstream hit compound confirmation and specificity assays. The specificity of the fluorescent H2 substrate in the RNase H2 cell lysate assay has been demonstrated previously, as no activity was detectable in mouse RNase H2 knockout embryo lysates 7 : recombinant RNase H2 was tolerant to 10% DMSO in the assay (data not shown), facilitating both single-point screening (1% final DMSO) and dose-response screening of compounds in up to 10% DMSO. Robustness of the high-throughput assay format was assessed by determining the Z′ factors for activator and inhibitor assay windows ( Fig. 1D ). In the absence of a known activator, recombinant RNase H2 enzyme at 2.5 U was used as a “high” assay control, and the Z′ was calculated from this under the assumption that an activator would result in a 2.5-fold increase in activity (under linear reaction conditions) relative to the 1 U concentration used in the HTS for assaying the compounds. The “low” assay control, “substrate alone” in which no RNase H2 enzyme was added was used similarly to define the inhibitory Z′ window. A mean activator Z′ of 0.77 and inhibitor Z′ of 0.88 were obtained, providing assay windows suitable for both activator and inhibitor compound screening ( Fig. 1D ). The 1 U RNase H2 concentration was therefore subsequently used to provide an assay window sufficient to allow identification of compounds either inhibiting or activating the RNase H2 enzyme.
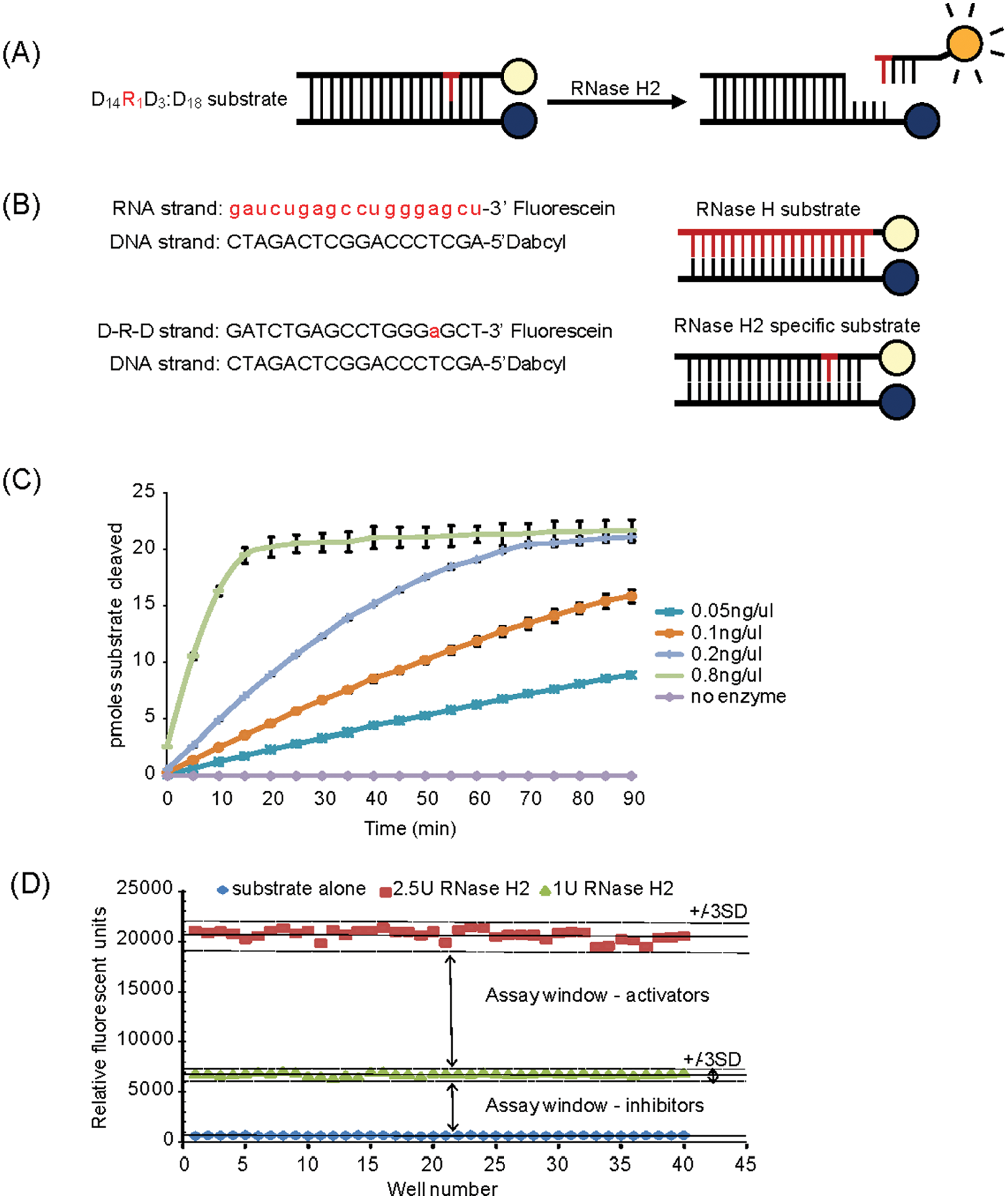
RNase H2 assay design. (
HTS Campaign and Hit Confirmation
The NINDS collection of 1040 compounds was used as pilot screen to validate the RNase H2 assay for HTS and establish a hit validation strategy (outlined in Fig. 2A ). A Z′ average of 0.62 was obtained for this pilot, with an initial hit rate of 3.5% using a <60% activity cutoff for the identification of inhibitors. Hit compounds were validated using a five-point dose response curve (DRC) against the recombinant RNase H2 and lysate RNase H2 assays, with low-micromolar IC50 activity achieved for 61% of initial hits, validating the HTS for RNase H2 inhibitor identification.
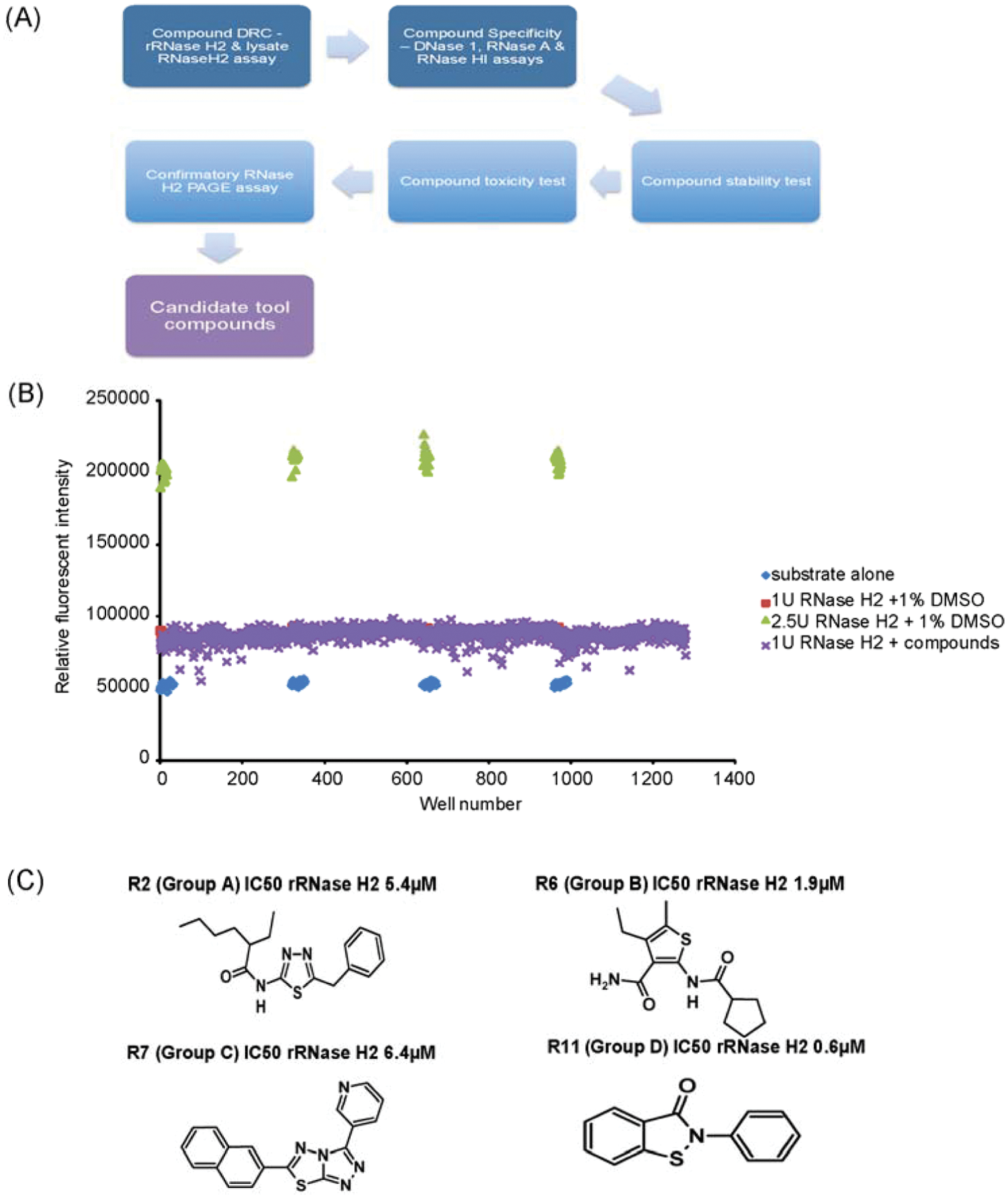
High-throughput screening (
A full HTS screen was then performed against 47 520 compounds. These comprised chemicals selected from diverse compound libraries from a variety of commercial suppliers, a library of kinase inhibitors, and a natural products library. Representative data from a single robotic HTS run screening four 384-well assay plates is shown in Figure 2B . With eight runs being completed per day, a throughput of 10 240 compounds/day was achieved. An initial hit rate of 1.3% was seen with the diversity and kinase libraries, whereas the natural product collection achieved a higher initial hit rate (10%). Forty-five inhibitor compounds were subsequently confirmed by RNase H2 DRC assays.
Activator compounds were not identified in the NINDS pilot and full HTS screen. All such initial HTS hits were subsequently eliminated as either autofluorescent compounds, active directly against substrate rather than the enzyme, or nonselective in downstream hit confirmation testing. Activators are likely to be much rarer compounds than inhibitors, and therefore screening of a much larger compound set will be required to identify such compounds. Also, the biological properties of RNase H2 may not be amenable to activation by small molecules. As well, a screen against recombinant RNase H2 containing patient mutations may be necessary to identify therapeutically relevant activators.
Four Classes of Small Molecules Have Potent RNase H2 Inhibitory Activity
The confirmed hits comprised four main families of related chemical compounds (groups A–D) as well as a number of structurally unrelated compounds (
Fig. 2C
). Twenty-four of these hits were selected for further study, along with five additional small-molecule compounds with structural similarity to the original hits. Two compounds identified in a previous study as inhibitors of HIV-1 RNase H were also purchased for comparative analysis, to establish if they were active against human RNase H2.
17
In total, 31 compounds were characterized in follow-up studies (data summarized in
Nineteen of these compounds had nM range IC50 activity against recombinant RNase H2 ( Fig. 3A ). To establish compound efficacy in a more physiologically relevant setting, dose-response testing against HeLa cell lysate containing RNase H2 activity was then performed. This identified two compounds (R11, R14) with nM activity ( Fig. 3B ), corresponding to the two group D compounds with the highest potency against recombinant RNase H2. A further eight compounds had activity between 1 and 10 µM, whereas 13 compounds showed IC50 >50 µM against RNase H2 activity in lysates, representing compounds with the weakest activity. Many group D compounds (the group with the highest RNase H2 activity) also demonstrated some activity against RNase HI ( Fig. 3C ), with micromolar IC50 values that ranged from 50× to 250× of those against RNase H2. Group B compounds had low µM activity against recombinant RNase H2 and lysate but no detectable activity against RNase HI, potentially suggesting higher specificity for RNase H2. Although group A, group C, and natural product compounds exhibited strong activity against recombinant RNase H2, they generally had poor activity in lysate enzyme assays and therefore appear less desirable reagents for cell biology tool compounds. Notably, the HIV RNase H compound group contained one compound with low µM IC50 against recombinant RNase H2, indicating cross-reactivity of some HIV RNase H inhibitors with endogenous RNase H enzymes. Such cross-reactivity may be of therapeutic relevance, and future counterscreening against human RNase H enzymes would be warranted to minimize cross-reactivity as a potential source of off-target toxicity by HIV-RT RNase H inhibitors.
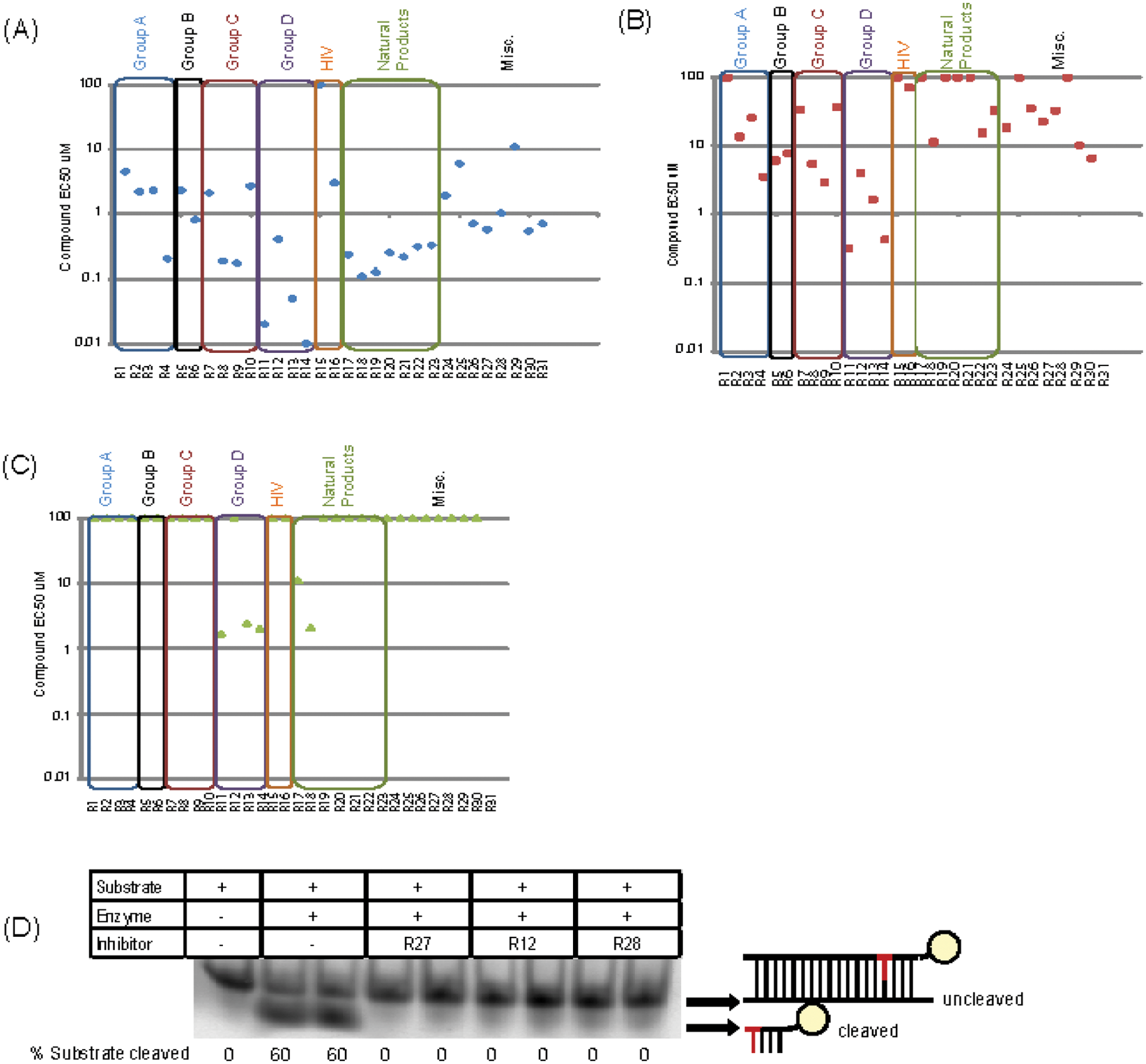
Hit validation. Nineteen compounds have nanomolar range inhibitory activity agents against recombinant RNase H2. (
A secondary assay was performed to ensure that compounds inhibited RNase H2 activity, rather than reducing fluorescence in the HTS assay by an indirect mechanism such as intercalation with the nucleic acid substrate. This secondary assay involved monitoring cleavage of the hybrid substrate by RNase H2 using polyacrylamide gel electrophoresis after incubation of a fluorescein-labeled substrate with test compound and RNase H2 enzyme (
Fig. 3D
). Most (21/31) small molecules entirely inhibited substrate cleavage under the assay conditions, confirming that they directly inhibit RNase H2 enzymatic hydrolysis (summarized in
To assess the potential utility of hits as pharmacologically useful compounds, the stability and cellular toxicity of the compounds were assessed (
The Group D Compound Structure Is a Potent Inhibitor of Ribonuclease H Activity
Follow-up studies established group D as a major class of compounds with high potency resulting in nanomolar-range activity in recombinant RNase H2 assays. Given that RNase H2 is the major RNase H in the cell,1,7 these compounds are likely to be potent general inhibitors of cellular RNase H activity. This suggests that they might be valuable tools for investigating the physiological roles of this class of enzyme in the cell. We therefore obtained seven additional compounds (R32–R38) with structural similarity to confirm the strong inhibitory activity for this structural group. These were characterized using the hit validation strategy, with one of the original and most active group D compounds (R14) used as a comparator (
Table 1
;
Fig. 4
;
Selected Group D Compounds, Structure and Inhibitory Activities.
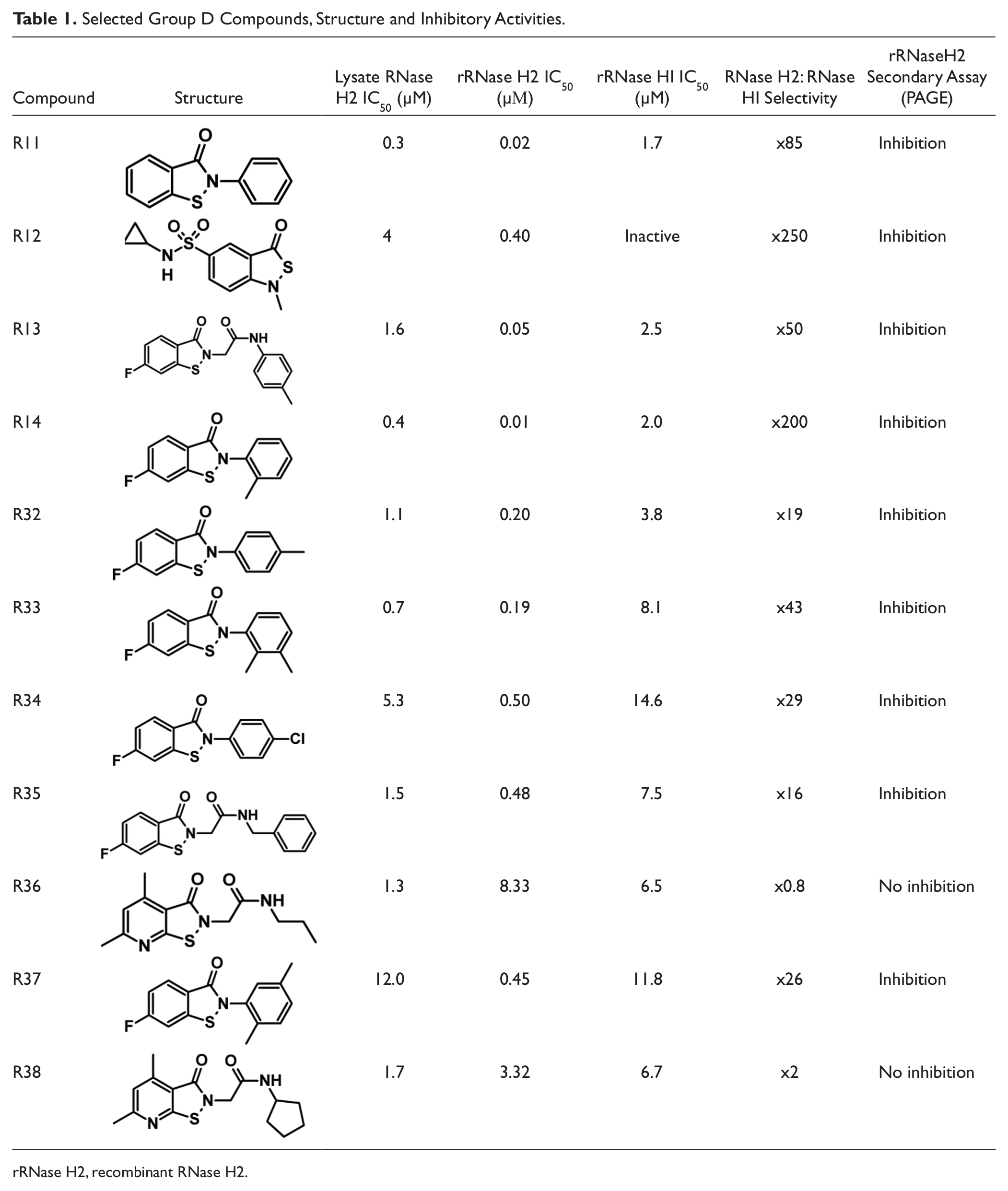
rRNase H2, recombinant RNase H2.
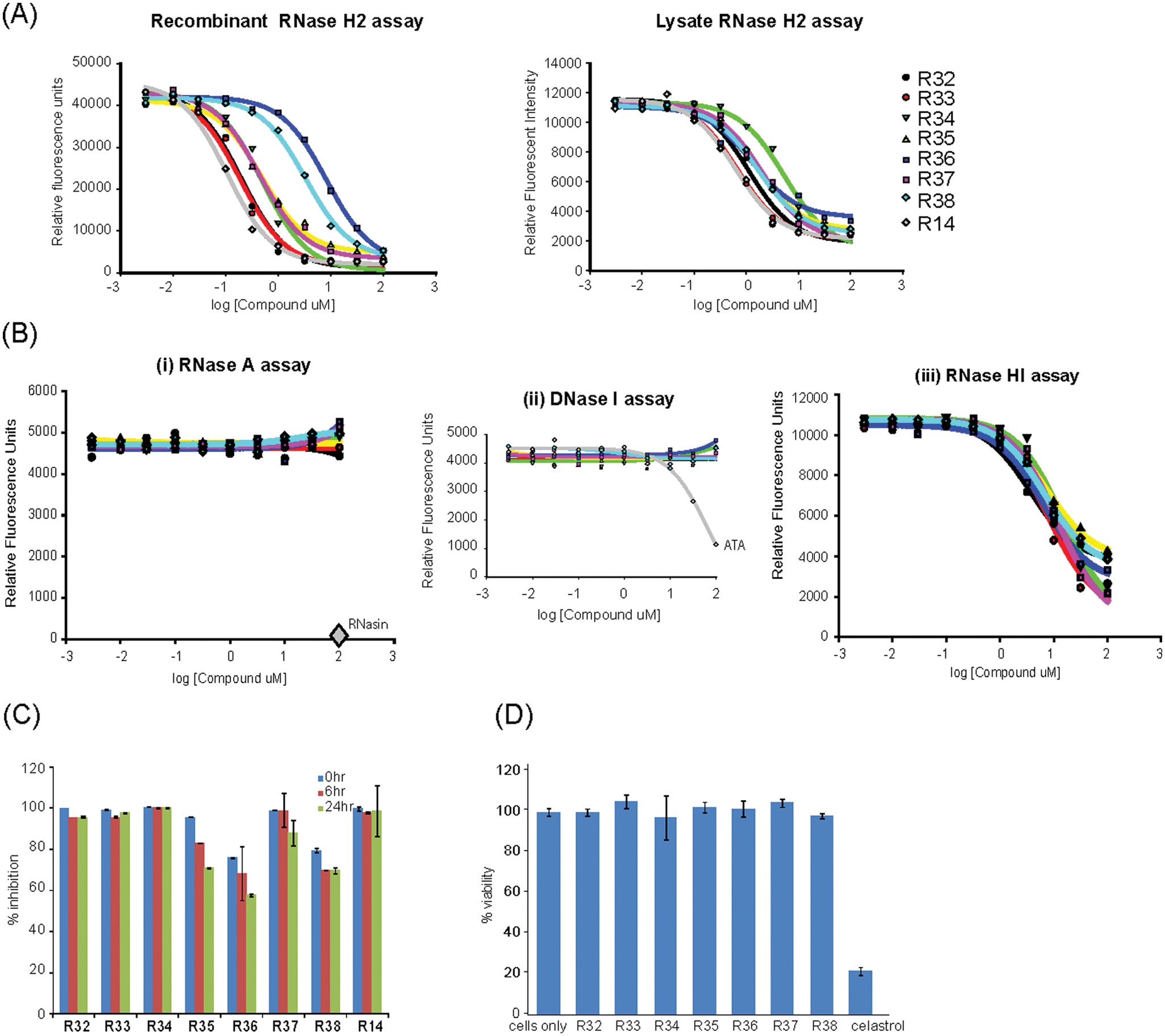
Group D compounds potently inhibit RNase H activity in vitro and in cells. (
In conclusion, group D compounds have been confirmed as a class of compounds showing potent inhibitory activity against RNase H2.
We propose compounds R14 and R11 from group D as the most promising candidates for tool compounds, which could be used to investigate the cellular function of RNase H enzymes. With potential for RNase H2 selectivity within the group being demonstrated with R14 and R12, structural refinement has the potential to create potent and highly selective RNase H2 inhibitors. Other classes of compounds (group B) have selectivity, showing specificity for RNase H2 with no activity against RNase HI.
Further optimization of the structures identified will be important to derive effective tool inhibitors for cellular studies. The current compounds are predicted to be membrane permeable but could be improved by substitution of additional polar chemical groups to reduce their lipophilic nature and enhance solubility. The reduced potency of compounds in the lysate assay may be due to increased environmental complexity, with the competition of other intracellular proteins also (nonspecifically) binding the inhibitors. This too may be improved by further structure-activity and selectivity refinement.
Establishing that these compounds are active in cells, at levels that do not cause cytotoxicity, will be key to demonstrating their utility as tools compounds. However, no assay currently exists to assess RNase H2 enzymatic activity in intact cells, and such assays may be technically challenging. Alternatively, a functional endpoint assay of RNase H2 deficiency may be used to demonstrate the cellular activity of inhibitors. This may become possible if sensitive assays are developed based on phenotypes identified in recently published knockout mouse models.7,8 Small-molecule inhibitors will be useful for the study of RNase H2 function, providing complementary approaches to conventional techniques such as RNAi. Small molecules have potential advantages over other approaches, such as reversibility of action and rapid temporal inhibition. Previous small-molecule screens have identified useful research tool compounds in the study of kinases20–22 and aldehyde dehydrogenases 23 and have been successfully used to study the cellular functions of their targets. Compounds identified in this study and their derivatives could therefore serve as useful reagents for future investigations into the cellular role and regulation of RNase H2.
Footnotes
Acknowledgements
We thank Puneet Khurana for help with the HTS robotics, Martin Reijns for advice and comments on the manuscript, Edward Jackson for proofreading, members of the Jackson Laboratory for useful discussion, and Craig Nicol for assistance with illustrations.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the MRC Technology Development Gap Fund. Work in the Jackson Lab is also supported by the MRC and by the Lister Institute of Preventative Medicine.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.