
Abstract
Detection and quantification of low doses of botulinum toxin serotype A (BoNT/A) in medicinal preparations require precise and sensitive methods. With mounting pressure from governmental authorities to replace the mouse LD50 assay, interest in alternative methods such as the endopeptidase assay, quantifying the toxin active moiety, is growing. Using internal collision-induced fluorescence quenching, Pharmaleads produced peptides encompassing the SNAP-25 cleavage site: a 17-mer (PL63) and a 48-mer (PL50) reaching the previously identified α-exosite, with PL50 showing higher apparent affinity for BoNT/A. Peptide mapping experiments revealed that this increased affinity is mainly due to a connecting peptide sequence between the N-terminus of PL63 and the α-exosite, identifying a new cooperative exosite on BoNT/A. Other endopeptidase substrates available, including SNAPTide and BoTest A/E, are both based on fluorescent resonance energy transfer (FRET) technology. To compare these assays, their limits of detection and quantification were determined using light chain or 150-kDa BoNT/A. Detection limits of PL50 and BoTest were over 100 times better than those using SNAPtide in standard conditions. Although the BoTest possessed a detection limit around 0.2 pM for either BoNT/A form, its quantification limit (9.7 pM) using purified BoNT/A was 12 times inferior to PL50, estimated at 0.8 pM, suitable for medicinal preparation quantification.
The botulinum neurotoxin (BoNT) family consists of seven antigenically distinct serotypes, BoNT/A–G, acting on the peripheral nervous system. 1 Among these toxins, serotypes A, B, E, and F can cause botulism in humans, a disease characterized by flaccid muscular paralysis, which, untreated, may lead to death, and BoNT/A is recognized as by far the most toxic. Its oral lethal dose 50 (LD50) for humans, estimated based on experiments using various animals, including monkeys, is 1 µg/kg. 2 Nevertheless, because of their neurological properties, very low doses of BoNT/A and B are widely used as therapeutic agents for the treatment of muscular and nerve disorders 3 and have also been shown to be efficient in the treatment of more than 30 different symptoms associated with neurological diseases. 4 Moreover, there is a substantial increase in the use of BoNT/A in aesthetics for wrinkle reduction. 5
The neurotoxins are produced in large quantities by the different types of anaerobic bacteria Clostridium botulinum as single inactive polypeptides of 150 kDa, which are subsequently processed by proteolytic cleavage into biologically active di-chains. 1 These active forms consist of an approximately 50-kDa light chain (LC) linked by a disulfide bridge to a 100-kDa heavy chain (HC) itself divided in two subdomains, designated as binding and translocation domains. The neurotoxins reach their intracellular targets by translocating the LC into the cytosol after endocytosis, via interaction of the HC with a high-affinity membrane-bound receptor complex such as the synaptic vesicle protein (SV2) for BoNT/A. 6 The LC, which possesses a highly specific zinc-endopeptidase activity, 7 then blocks synaptic vesicle fusion with the presynaptic membrane by selectively cleaving one of the three SNARE polypeptides involved in neuroexocytosis. BoNT/A, for instance, specifically cleaves the 206–amino acid SNAP-25 protein exclusively at the Q197R198 scissile bond, thus inhibiting neurotransmitter release at the neuromuscular junction.8,9
Procedures to be used for BoNT detection and quantification in toxin preparations for medical and aesthetics applications or in the event of malevolent bioterrorist acts have to be highly sensitive as well as selective. The advantage of the currently used pharmacotoxicological mouse LD50 assay, considered the gold standard assay, is to provide the in vivo toxicity of a given botulinum toxin sample whatever the nature of the infected medium. However, this assay is time-consuming, requires the use of a large number of animals, and has poor repeatability due to the large number of fluctuant parameters involved in this method. 10 Several in vitro assays have been reported for the detection and quantification of BoNT/A. These assays rely on mass spectrometry,11,12 immunological detection,13,14 or endopeptidase activity against the entire SNAP-25 or fragments thereof.15–17 The advantage of the latter assay is that it measures and quantifies the active component of the toxin, which is directly responsible for its toxicity. With mounting pressure from the authorities to reduce animal consumption, use of endopeptidase assays in production lines for quality control purposes could prove highly advantageous, insofar as the assay is sensitive enough to detect low doses of toxin.
On the basis of the SNAP-25 sequence, we developed a short peptide substrate for BoNT/A called PL63 ([SNAP-25(Ac-187-203)] (Q/Nop197, T/Pya200, M/Nle202)). It is fluorescently quenched by internal collision-induced fluorescence quenching (ICIFQ) 18 after the introduction of the fluorophore/repressor pair made of the highly fluorescent pyrenylalanine (Pya) along with the nitro-phenylalanine (Nop) repressor residue in the Q197 and T200 positions. 19 Although this substrate was well recognized and cleaved by BoNT/A, peptide mapping experiments were performed in this study to investigate whether extending its sequence to the previously identified cooperative binding α-exosite 20 could significantly improve its enzymatic specificity. Results of these studies bring to light a new cooperative exosite for BoNT/A native substrate binding.
Based on these experiments, PL50 ([SNAP-25(Ac-156-203)] (Q/Nop197, T/Pya200, M/Nle202)) was synthesized and compared with other available BoNT/A substrates. Although all commercially available endopeptidase assays rely on peptide substrates based on the SNAP-25 sequence, they are of varying lengths and use different techniques of fluorescence. Indeed, SNAPtide is a short peptide with a fluorescent dye donor moiety at one end and a fluorescent resonance energy transfer (FRET) quencher at the other. Based on the fluorescent sensors published by Dong and collaborators, 21 the now commercial BoTest A/E uses true FRET, with the SNAP-25(141–206) sequence being flanked by the cyan (CFP) and yellow (YFP) fluorescent protein donor-acceptor pair. Establishing the limits of detection and quantification of PL50, SNAPtide, and BoTest A/E suggests that longer peptides such as BoTest A/E and PL50 provide better sensitivity but also reveals that the FRET BoTest A/E reporter may not be appropriate for in vitro enzymatic assay. Taken together, the results show that the PL50 assay allies linearity, high sensitivity, and specificity toward BoNT/A, being the single assay able to detect less than 50 pg of 150 kDa BoNT/A.
Materials and Methods
Materials
Recombinant light chain, purified 150 kDa BoNT/A, and SNAPtide (FITC/DABCYL) were from List Biological Laboratories (Campbell, CA), and the BoTest A/E reporter (CFP–SNAP-25141–206–YFP) was purchased from BioSentinel (Madison, WI). HEPES, ZnSO4 (50 mM solution), dithiothreitol (DTT), trimethylamine N-oxide (TMAO), and tris(2-carboxyethyl) phosphine (TCEP, 0.5 M, pH 7) were from Sigma (Lyon, France), and Tween 80 was purchased from Fluka (Basel, Switzerland). Fmoc amino acids and coupling reagents were purchased from Applera France (Courtaboeuf, France). Fmoc-L-(4-nitro)-phenylalanine was from Bachem Distribution (Weil am Rein, Germany) and Fmoc-L-pyrenylalanine was from PolyPeptide (Strasbourg, France). HMP and MBHA resins were from Novabiochem (VWR, Fontenay sous Bois, France), and solvents were from Carlo Erba-SDS (Vitry, France). Half 96-well low-binding black plates were from Corning (Corning, NY).
Peptide Synthesis
Peptides were synthesized by the solid-phase method, using an ABI 433A automated synthesizer (Applied Biosystems, Foster City, CA), coupled to a 785A programmable absorbance UV detector, based on classical Fmoc chemistry with HBTU/HOBt/DIEA as coupling reagents, as described in Poras et al. 17 Purity of the peptides, determined by high-performance liquid chromatography (HPLC), was greater than 98.5%, and their molecular weights (MW) were confirmed by deconvolution of the mass spectra obtained with an electrospray mass spectrometer using Xcalibur software (Thermo Finnigan, San Jose, CA) (see supplemental data for details concerning each peptide):
PL63 and PL50 Kinetic Parameters
Kinetic parameters were evaluated using fluorescent methods. For this purpose, BoNT/A LC at either 50 ng/mL for PL63 or 10 ng/mL for PL50 was diluted in reaction buffer (20 mM HEPES [pH 7.4], 200 µM ZnSO4, 5 mM DTT, 1 mg/mL bovine serum albumin [BSA]) with increasing concentrations of either substrate. Reactions with PL63 and PL50 were incubated at 37 °C for either 30 or 10 min, respectively. Fluorescence was read on a Berthold Twinkle (Berthold Technologies, Bad Wildbad, Germany) fluorimeter (λex = 340 nm, λem = 405, lamp energy = 10 000). Calibration curves connecting the measured increase in fluorescence intensity to changes in the molar ratio of substrate/metabolite were established in assay buffer by mixing increasing concentrations of fluorescent metabolite and related decreasing concentrations of substrate.
Fluorescent and HPLC Analysis of Peptide Cleavage and Impact of Exosite-Binding Peptides
Peptides corresponding to the previously identified α-exosite (SNAP-25 aa152–168) and to the connecting peptide (SNAP-25 aa169–186) were added in excess (20 times), alone or in combination, to BoNT/A LC toxin (50 ng/mL) in 20 mM HEPES buffer (pH 7.4) containing 5 mM DTT, 0.2 mM ZnSO4, and 1 mg/mL BSA and preincubated at 37 °C for 30 min, whereupon PL63 substrate (10 µM) was added and further incubated for 60 min at 37 °C. At the end of the incubation, the reactions were stopped by the addition of 20 µL 1,4-dioxane and loaded onto a Kromasil C18 column (5 µm, 100 Å, 4.6 × 250 mm). The hydrolysis products were separated using a 0% to 60% CH3CN (0.1% trifluoroacetic acid [TFA]) gradient in 30 min with a flow rate of 1 mL/min. The detection was performed by UV at 343 nm, characteristic of the Pya moiety. Peaks corresponding to the fluorescent metabolite, PL100, were integrated and quantified using a scale run in the same HPLC conditions. Negative control experiments were performed in parallel using nonrelated peptides, substance P and bradykinin (Bachem Distribution, Weil am Rhein, Germany), in the same conditions. Kinetic parameters of PL63 alone or combined with the α-exosite and connecting peptides were also evaluated using the same HPLC method and toxin concentrations ranging from 0 to 50 ng/mL.
Standard BoNT/A LC Toxin Detection Curves
PL63 assay
Standard curves were obtained using decreasing concentrations of BoNT/A LC diluted in reaction buffer (20 mM HEPES [pH 7.4], 200 µM ZnSO4, 5 mM DTT, 1 mg/mL BSA) and preincubated for 15 min at 37 °C. PL63 was solubilized in 20:80 DMF/H2O (v/v) at 1 mM and diluted to a 200-µM working solution in reaction buffer. After the preincubation time, PL63 was added to a final concentration of 10 µM in a final volume of 100 µL and the reaction left to proceed for 5 h at 37 °C. The fluorescent signal was read on a Berthold Twinkle fluorimeter (λex = 340, λem = 405, lamp energy = 10 000). This experiment was performed three times independently in duplicate.
SNAPtide (FITC/DABCYL) assay
Standard curves were obtained using increasing concentrations of BoNT/A LC toxin diluted in HEPES 20 mM (pH 7.4) and Tween 0.05% reaction buffer and preincubated at 37 °C for 15 min. SNAPtide (FITC/DABCYL) was suspended according to the supplier’s instructions and diluted in reaction buffer. After the preincubation period, the reaction was begun by addition of the SNAPtide substrate (final concentration 8 µM) in a final volume of 100 µL and the reaction left to proceed for 5 h at 37 °C. Fluorescence was read on a Berthold Twinkle fluorimeter (λex = 485 nm, λem = 535 nm, lamp energy = 10 000). This experiment was performed three times independently in duplicate.
PL50 assay
Standard curves were obtained using decreasing concentrations of BoNT/A LC diluted in optimized reaction buffer (20 mM HEPES [pH 7.4], 200 µM ZnSO4, 2.5 mM TCEP) and preincubated for 15 min at 37 °C. PL50 was solubilized in 20:80 DMF/H2O (v/v) at 1 mM and diluted to a 200-µM working solution in reaction buffer. After the preincubation time, PL50 was added at a final concentration of 10 µM in a volume of 100 µL and the reaction left to proceed for 5 h at 37 °C. Using this substrate, the same experiments were also performed using purified 150 kDa BoNT/A. Decreasing concentrations of 150 kDa BoNT/A were preincubated in reaction buffer so as to release the active LC chain from the 150-kDa protein through the action of TCEP. The fluorescent signal was read on a Berthold Twinkle fluorimeter (λex = 340, λem = 405, lamp energy = 10 000). These experiments were performed three times independently in duplicate.
BoTest A/E reporter assay
Dose-response curves were obtained using decreasing concentrations of BoNT/A LC in reaction buffer (50 mM HEPES [pH 7.1], 5 mM NaCl, 1% Tween-20, 100 µM ZnSO4) and preincubated at 37 °C for 15 min. The reaction was started by the addition of the BoTest A/E peptide reporter at the recommended final concentration of 10 µM. Incubation times were 5 h at 37 °C (final volume 100 µL), after which time the fluorescent signal was measured on a Berthold Twinkle fluorimeter (λex = 430, λem = 460 and 535, lamp energy = 10 000) and the ratios calculated. The same experiments were performed with purified 150 kDa BoNT/A by adding a 5-mM concentration of DTT, as recommended by the manufacturer, to the reaction buffer to release the BoNT/A LC. These experiments were performed three times independently in duplicate.
Limit of Detection and Quantitation Determinations
The limit of detection (LoD) of a BoNT/A assay corresponds to the lowest detectable toxin concentration using one particular assay in defined conditions. This measure is highly dependent on the background signal of a given substrate, and it can be estimated using the following formula: LoD = mean + 3SD, in which “mean” corresponds to the mean value of the signal produced by the substrate diluted in reaction buffer, and SD is the standard deviation from that mean signal.21,22
Thus, repeated measures of each substrate diluted to its final assay concentration in its corresponding reaction buffer and incubated at 37 °C were performed (i.e., 10 µM PL63 in 20 mM HEPES [pH 7.4], 200 µM ZnSO4, 5 mM DTT, 1 mg/mL BSA reaction buffer; 8 µM SNAPTtide in HEPES 20 mM [pH 7.4], Tween 0.05% reaction buffer; 10 µM BoTest A/E reporter in 50 mM HEPES [pH 7.1], 5 mM NaCl, 1% Tween-20, 100 µM ZnSO4; and 10 µM PL50 in 20 mM HEPES [pH 7.4, 200 µM ZnSO4, 2.5 mM TCEP). Fluorescence was read as described above for each substrate. The calculated LoD values were then reported on their corresponding standard curves to obtain the lowest toxin concentration detectable using that assay.
The limit of quantitation (LoQ) of an assay is the lowest toxin concentration that can be quantified with acceptable precision and accuracy under the stated conditions of the given assay.21,22 Although there are many ways to determine an LoQ, in the statistical method, its value is set at 10 SD above the mean blank value. Assay LoQs were calculated using this method and reported on their corresponding standard curves to obtain the lowest quantifiable toxin concentration for each assay.
Effect of TMAO on Peptide Substrates
The effect of the osmolyte TMAO was studied by the addition of a final concentration of 1 M to each reaction buffer. Its effect on PL63 was studied using 10 ng/mL BoNT/A LC after a 3-h incubation at 37 °C in reaction buffer (20 mM HEPES [pH 7.4], 200 µM ZnSO4, 5 mM DTT) with or without BSA. The effect of the osmolyte on PL50 cleavage was studied in the same conditions, without BSA. For experiments using SNAPtide, after an initial experiment using 10 ng/mL BoNT/A LC incubated 3 h at 37 °C, a full scale was performed as described above but in the presence of the osmolyte at 1 M. For the BoTest A/E, the effect of the addition of TMAO 1 M to the supplied buffer was studied over a range of toxin concentrations, to produce a standard curve. End-point fluorescence readouts were performed after 5 h at 37 °C, as described above for each assay. Deltas of fluorescence were calculated by subtracting the base fluorescence of the peptides incubated in parallel in buffer alone for the PL63, PL50, and SNAPtide substrates, and ratios were calculated for the BoTest A/E.
Data Manipulation and Statistical Analysis
All data were analyzed using GraphPad Prism 4 (GraphPad Software, La Jolla, CA), and statistical analyses were performed using the Student t test.
Results
To develop a highly sensitive enzymatic assay, we first designed a fluorescently quenched peptide of 17 amino acids called PL63. This short peptide covered amino acids 187 to 203 of the SNAP-25 sequence, with the quencher moiety (S)-4-nitrophenylalanine (Nop) replacing Q197 and the extremely fluorescent (S)-pyrenylalanine (Pya) moiety in position 200, replacing the natural threonine residue at this position. Study of the kinetic parameters of this substrate toward BoNT/A LC revealed that it binds the toxin with an affinity of 14.5 ± 0.9 µM, whereas its catalytic efficiency (kcat/Km) was of 1.40 ± 0.06 105 M–1·s–1 ( Fig. 1A ). Using this quenched substrate, BoNT/A limits of detection and quantitation (LoD and LoQ) were determined to be 1 and 3 ng/mL, respectively ( Fig. 1B ). Thus, although this substrate may be useful for inhibitor screening, it appears not sensitive enough to detect and quantify the low toxin concentrations (ranging from 100–500 mouse LD50s or 200 pg to 1 ng) used in medical formulations.
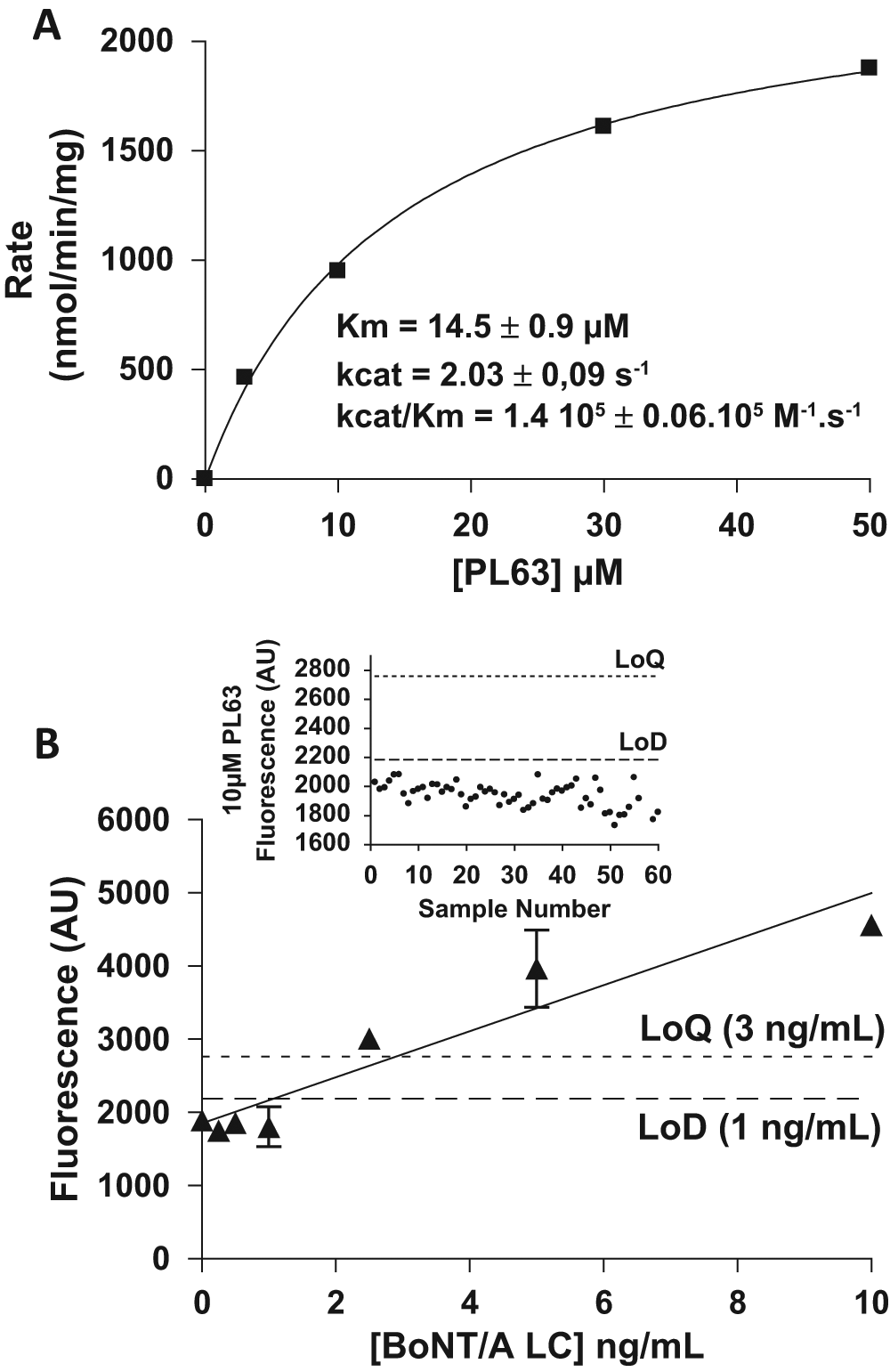
(
The unrivaled specificity of tetanus and clostridial neurotoxins has been attributed to the binding of multiple exosite sequences, remote from their catalytic sites.20,23 In BoNT/A, binding to the α-exosite sequence has been shown to be essential for proper substrate orientation and optimal catalytic activity. 20 To ascertain the positive effect of extending the sequence of PL63 to the α-exosite binding sequence, we performed peptide mapping experiments. For this purpose, two peptides were synthesized, one corresponding to the previously identified α-exosite sequence (Ac-152QVSGIIGNLRHMALDMG168) and the second (169NEIDTQNRQIDRIMEKAD186) to the sequence separating the latter from the N-terminus of the PL63 substrate, refered to as the “connecting peptide.” The effect of their addition on the cleavage of PL63 as well as on its kinetic parameters was monitored by HPLC. Results of these experiments, presented in Figure 2 , show that the addition of the α-exosite binding sequence did increase the cleavage of PL63. Moreover, the connecting peptide alone was found to have an even greater cleavage-promoting effect (33% increase with connecting peptide vs. 18% with the α-exosite alone). The greatest impact on PL63 cleavage (56%) was observed by their concomitant addition (while the addition of two unrelated peptides had no effect; not shown), suggesting that both sequences may bind exosite amino acid sequences on the LC. This increased efficiency of the LC BoNT/A toxin was further confirmed by the study of the kinetic parameters of PL63 alone or in the presence of the exosite-binding sequences, which revealed that their addition endowed PL63 with a lower Km value, from ~19 µM without to 11 µM with the exosites ( Fig. 2 ). Moreover, as these latter experiments were performed by HPLC, they also confirm the kinetic parameters measured using fluorescence.
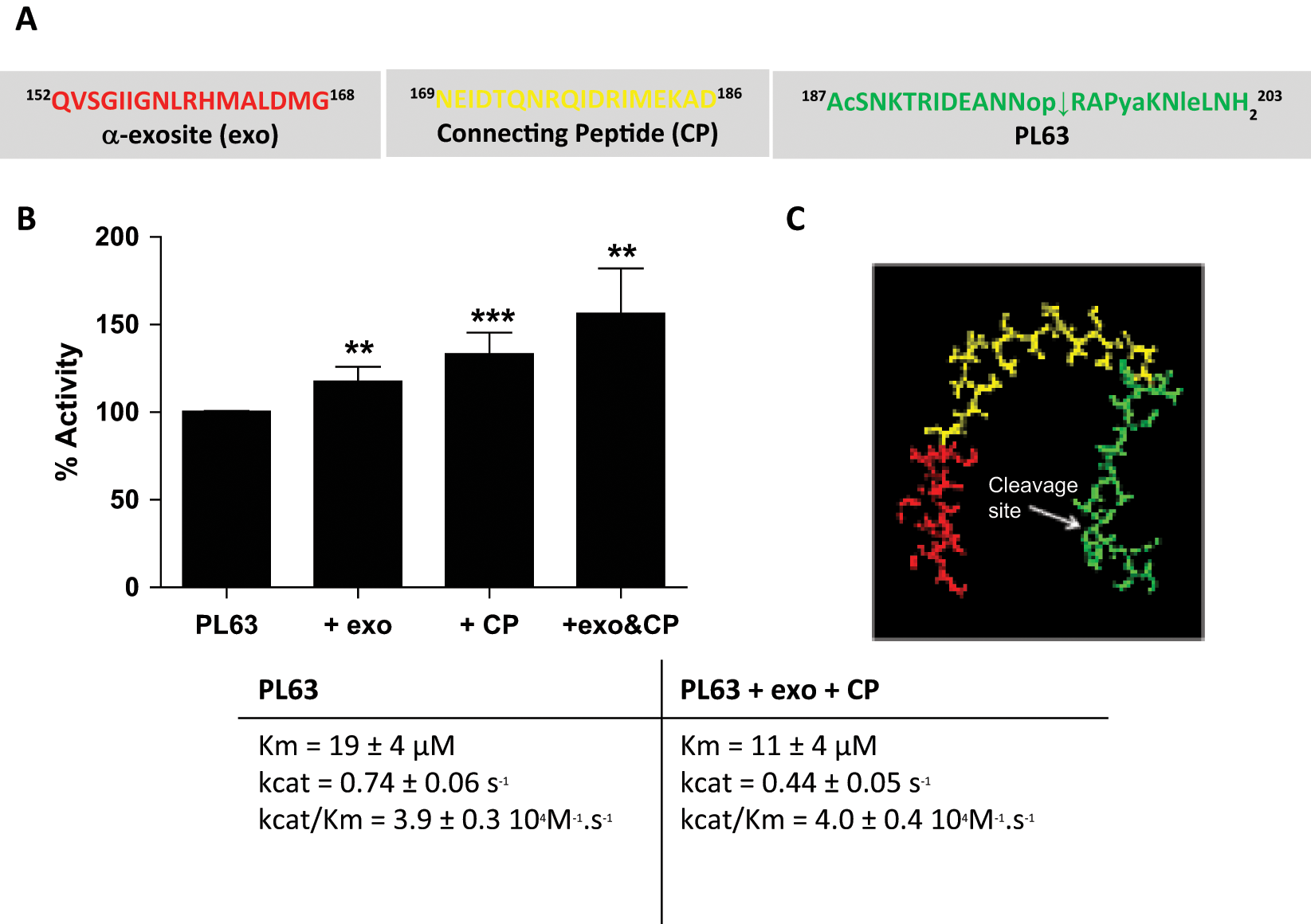
(
On the basis of these results, we synthesized a longer peptide substrate covering amino acids 156 to 203 of the SNAP-25 peptide sequence (i.e., the sequence of PL63 linked to the connecting and α-exosite sequences; Fig. 2 ). Determination of the enzymatic constants of this longer substrate designated PL50 showed that it binds the BoNT/A LC with a Michaelis-Menten constant (Km) of 0.8 ± 0.21 µM, increased almost 20-fold as compared with PL63. Although the catalytic rate of PL50 was not significantly modified, its resulting catalytic efficiency (5.4 ± 0.4 106 M–1·s–1) was increased by more than 40-fold ( Fig. 3A ). The LoD and LoQ of the PL50 assay in optimized conditions (50 and 270 pg/mL, respectively) also decreased by 20- and 11-fold, respectively, as compared with PL63 ( Fig. 3B ). The highly sensitive PL50 assay also remained linear over a wide range of toxin concentrations, as displayed in Figure 3C .
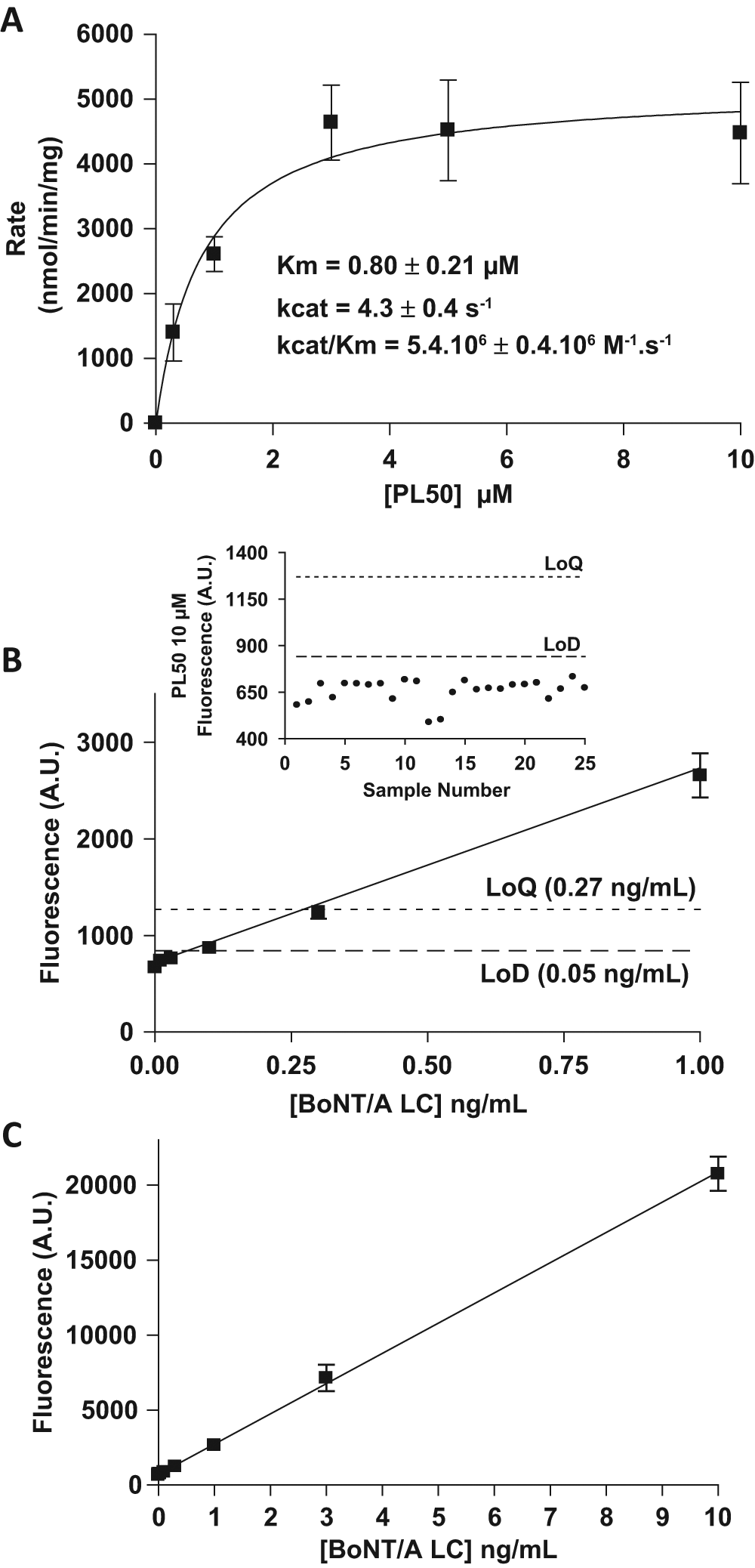
(
Having designed a BoNT/A assay able to detect only a few picograms of toxin, its LoD and LoQ were compared with the two other endopeptidase assays available on the market (i.e., SNAPtide and BoTest A/E). The LoD and LoQ of SNAPtide were estimated at 10 and 37.5 ng/mL using an incubation time of 5 h in the described conditions ( Fig. 4A ). The high LoQ measured for SNAPtide was the result of the high and variable background fluorescence of this substrate. The BoTest A/E detected much lower concentrations of LC, 10 pg/mL (0.28 pM), although its LoQ was 370 pg/mL for BoNT/A LC ( Fig. 4B ).
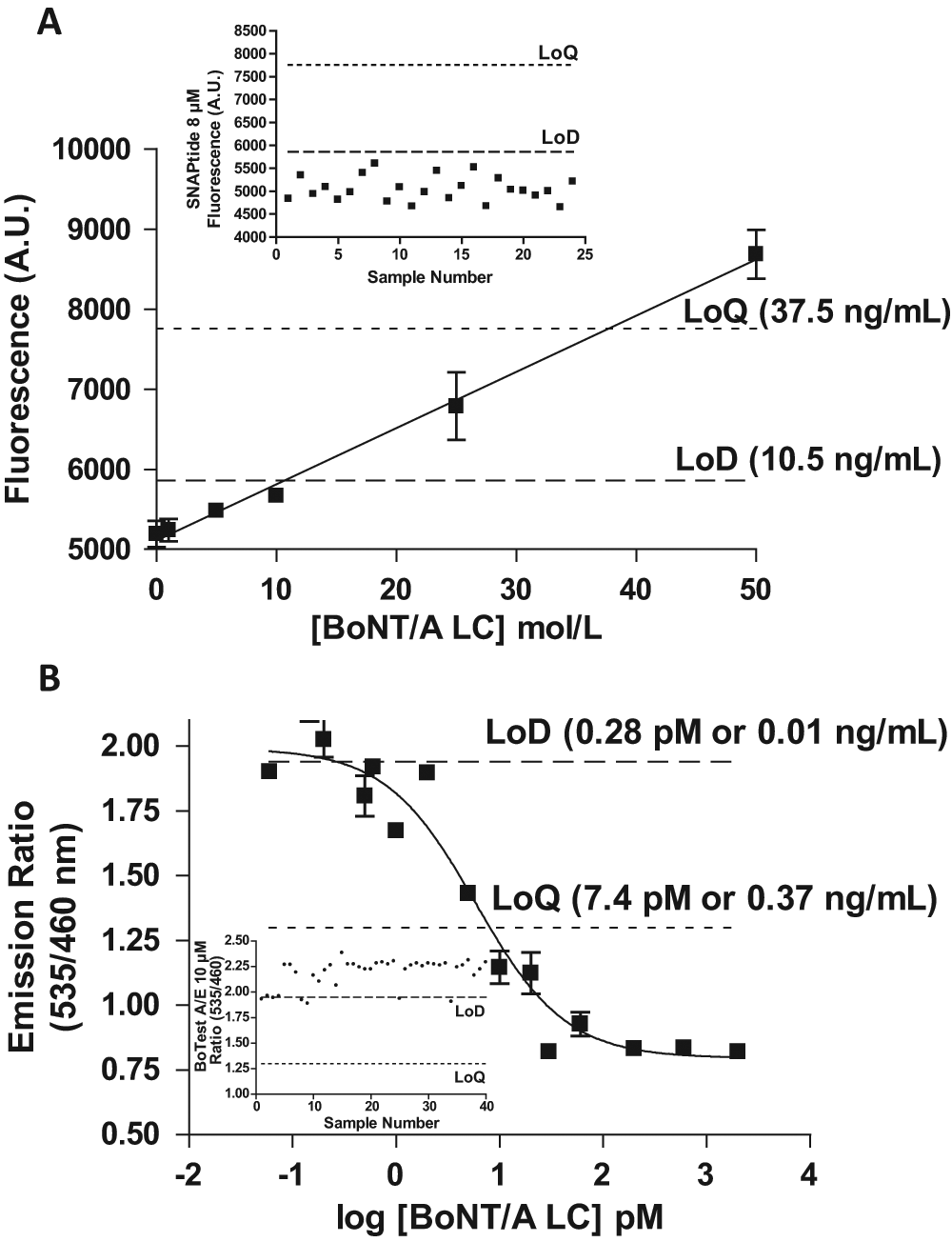
Compared limits of detection (LoDs) and limits of quantitation (LoQs) of SNAPtide and the BoTest A/E: (
Osmolytes are small organic compounds known to affect protein stability. 24 The effect of one such stabilizing agent, TMAO (1 M), on the activity of BoNT/A LC was investigated using the different substrates. Results of these experiments show that the effect of the osmolyte is dependent on the substrate used to assess the toxin enzymatic activity. Thus, whereas it did increase the cleavage of PL63 (in the absence of crowding agent BSA; Fig. 5A ) and that of the short commercial SNAPtide FRET peptide ( Fig. 5B ), it had a negative impact on the cleavage of the PL50 substrate ( Fig. 5C ). Interestingly, when the standard PL63 buffer, which includes 1 mg/mL BSA, was used, the impact of TMAO addition was no longer apparent. Using the BoTest A/E FRET reporter, the most significant effect of TMAO was on the reporter’s own conformation, as evidenced by the change in FRET measured at low toxin doses ( Fig. 5D ). As a result of this effect, the appearance of the sigmoidal standard curve was modified, with a shortened inflection zone, which was not advantageous to the assay.
Effect of trimethylamine N-oxide (TMAO) on the cleavage of peptide substrates by botulinum toxin serotype A (BoNT/A) light chain (LC). Illustrated in
The LoQ of the two most sensitive assays was further compared using purified 150 kDa BoNT/A. These results, illustrated in Figure 6 , confirm their high detection capacities, with the PL50 assay displaying an LoD of 50 pg/mL (0.33 pM) and the BoTest A/E of 23 pg/mL (0.15 pM). Although the LoD of the BoTest was superior to that of the PL50 assay, the 430 pg/mL (2.9 pM) LoQ of the latter was lower than that of BoTest A/E (LoQ = 1.46 ng/mL or 9.7 pM). Moreover, these results further confirm the linearity of the PL50 assay over a wide range of toxin concentrations ( Fig. 5B , inset), thus allowing, with a single scale, the detection and quantification of many different toxin concentrations.
Compared limits of detection (LoDs) and limits of quantitation (LoQs) of purified 150 kDa using the A/E BoTest or PL50: BoTest (
Discussion
Recently, we used internal fluorescence quenching to design novel substrates for the simple and sensitive assay of BoNT/A activity. 17 Of these, the fluorigenic PL63 is a short, 17-mer peptide (Ac-187-203 SNAP-25-NH2 (Nop197, Pya200, Nle202)) based on the sequence of its highly selective SNAP-25 substrate, whereas PL50 is a 48-mer peptide, corresponding to amino acids 156 to 203 (Ac-156-203 SNAP-25-NH2 (Nop197, Pya200, Nle202)). Both fluorigenic peptides encompass the natural cleavage site of SNAP-25. As previously shown using purified BoNT/A, increasing the length of PL63 to reach the α-exosite binding sequence, as in PL50, increases substrate binding. This is clearly evidenced here using LC BoNT/A, revealing a 20-fold increase in the Km value for the longer peptide substrate. Nevertheless, the fluorescent signal produced by the cleavage of PL63 after a short incubation time (≤60 min) is largely sufficient to perform high-throughput screenings of BoNT/A inhibitors (see Fig. 1A ). This substrate therefore provides both a rapid and cost-effective assay.
Cooperative hydrolysis of clostridial neurotoxin has previously been shown for TeNT 23 and has also been proposed for BoNT/A. 20 This phenomenon very likely plays a role in the exquisite selectivity of clostridial neurotoxins toward their substrate. In this study, the existence of such a mechanism in BoNT/A was explored by using a “minimal” substrate in the presence of two peptides corresponding to the binding sequences of the α-exosite and connecting peptide in SNAP-25. HPLC analysis demonstrated an improvement of the hydrolysis ( Fig. 2B ), although less important than for TeNT. 23 Using the peptide combinations, not only was the cooperative role of the α-exosite demonstrated, but the “connecting peptide” alone was also shown to have an effect, even greater than that of the α-exosite binding sequence. This result complements previously published data by Chen and Barbieri, 25 who had identified, using SNAP-25 mutants, the SNAP-25(167–181) region (B region) as being important for substrate binding to BoNT/A LC. With this B region corresponding to our connecting peptide, which spans amino acids 169 to 186 of the SNAP-25 protein, this SNAP-25 sequence is thus demonstrated as an additional cooperative site for BoNT/A.
Although the peptide mapping experiments allowed the delineation and comparison of the respective effects of each exosite, the optimal cleavage was observed with one single, longer peptide, PL50 (Ac-156-203 SNAP-25-NH2 (Nop197, Pya200, Nle202)), joining PL63, the connecting peptide and the α-exosite binding sequences. Indeed, with this 48-mer substrate, the Michaelis-Menten constant was very significantly decreased, almost 20-fold, as compared with PL63, most likely reflecting better substrate recognition.
Taken together, these results also explain why longer peptides, which bind all the necessary cooperative sites, possess better specificity constants than shorter peptides and thus better neurotoxin detection limits. This fact is further illustrated in Figure 2C , which displays, based on the co-crystal structure of BoNT/A LC with SNAP-25, 20 the structure possibly adopted by the three peptides constituting PL50. Indeed, the representation shows that the three- peptide combination may reach the horseshoe conformation observed in SNAP-25 promoted through toxin exosite binding. Exosite binding on the surface of the toxin is even more likely in the presence of a single, longer peptide substrate such as PL50. On the contrary, with shorter peptides such as PL63 or SNAPtide fitting only the binding sites surrounding the catalytic pocket of the toxin, the same conformational adaptation is never promoted, and thus, the peptide cleavage is diminished. Although the interaction of these shorter peptides with the BoNT/A LC can be favored by the use of osmolytes such as TMAO or crowding agents such as BSA, as demonstrated here ( Fig. 5 ) and also previously shown by Nuss and collaborators, 26 these additives do not induce the specific protein conformational modifications necessary for optimal peptide cleavage. This is also the case with the peptide combinations used in the peptide mapping experiments. Thus, with shorter peptides such as SNAPtide, although the BoNT/A LC detection limit (LoD) in the presence of TMAO (1 M) is decreased 3-fold from 10 ng/mL to 3.5 ng/mL ( Fig. 5C ), it remains far from the picomolar concentrations detected with both the BoTest A/E and the PL50 substrates, in buffers devoid of osmolytes or crowding agents ( Figs. 3B , 4B , and 6 ).
Comparing the performances of the PL50 assay with those of SNAPtide and BoTest A/E revealed that although the best detection limit was obtained using the BoTest A/E, its quantification limit, estimated at 1.46 ng/mL (or 146 pg per assay) for purified 150 kDa toxin, was inferior to that reached with PL50 (430 pg/mL or 43 pg per assay). These LoD values strongly suggest that only PL50 is suited for the quantification of medical preparations of BoNT/A, which contain very low concentrations of toxin. Indeed, BoNT/A vials used in aesthetics or for medical treatment often contain as low as 50 or 100 mouse LD50s, which can be estimated to correspond to ~100 or ~200 pg of toxin, respectively (with 1 mouse LD50 = 2 pg 17 ). Although both the BoTest A/E and the PL50 assay can detect these low toxin concentrations, only the PL50 assay provides a means to quantify these toxin preparations, even allowing for replicates in the case of 100 mouse LD50 formulations. Moreover, the BoTest A/E suffers from the FRET technology, which is more appropriate for peptide interaction or structural investigations than for proteolytic activity studies. Indeed, the substrate in its uncleaved form emits a fluorescent signal that is modified by energy transfer not only after cleavage but also as a result of intrinsic conformational changes, as evidenced by the effect of TMAO. The mathematical transformation of the signal combined with the logarithmic concentration scale also makes the test less precise than the ones measuring a direct signal over a linear scale (e.g., PL50, PL63, and SNAPtide). Another consequence of the nature of the BoTest A/E assay is that the linear part of the assay spans over an extremely narrow range of toxin concentration, requiring several dilutions of any tested material to find the appropriate, quantifiable one. Finally, the A/E reporter of the BoTest is less selective than the PL50 substrate.
Comparing the SNAPtide and PL50 assays reveals that, although assay optimization did significantly increase the detection and quantification limits of the former ( Fig. 5 ), PL50 remains the most sensitive as well as the simplest and specific BoNT/A assay for the detection and quantification of toxin preparations. With the linearity of the PL50 assay being observed over a broad range of toxin concentrations, any toxin preparation or formulation can easily be quantified, with an estimated LoQ of 0.43 ng/mL (i.e., 43 pg/well) for the purified 150 kDa BoNT/A.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.