
Abstract
The unmet need for the treatment of disorders of the nervous system is growing, and as highlighted in the media and elsewhere, the results of an aging population will ensure this continues with an upward trajectory. Incredibly, the efforts within industry to identify new drugs to treat these conditions have seemingly disappeared despite the growing need. There has been a run of extraordinary failure in the later stages of the drug discovery process for neurological and psychiatric disorders, which has many causes. We believe, though, that we have to confront this dire situation, both by using learnings from the post hoc analysis of our historical failure, as well as harnessing the bewildering array of new technologies and data now available to us, to ensure we are making the right decisions along the very complicated path of drug discovery to registration.
Introduction
The origins of the modern approach to CNS drug discovery arose from the desire to find more selective agents with less adverse side effects than the clinically efficacious compounds that were serendipitously discovered in the 1950s to 1970s for neuropsychiatric diseases. 1 This has led to a long and winding road of “me too” drugs, which were simply tweaks on older drugs but with reduced propensity for side effects.2–4 Although this “me too” approach was sustainable for a period of time, we are now in a period when such drugs are losing patent exclusivity and many pharmaceutical companies are dealing with the resultant “patent cliff.” 5 To rebound from this difficult period, many companies are significantly downsizing to manage costs. We believe that the appropriate solution, particularly in the area of CNS research, is to overhaul the strategy by which we identify, value, and validate new targets and develop them as drug discovery efforts. This will deliver novel differentiated medicines and not just another “me too.”
This review attempts to provide a devil’s advocate view of the traditional approach of CNS drug discovery, from the early target ID stages through evaluation in preclinical assays into aspects of clinical trial design, and proposes a paradigm shift for moving forward beyond the past decade(s) of clinical failures. We propose a future approach that will require a deep understanding of the biology and function of the target in normal and disease states and an appreciation of the drug’s mechanism of action.
The Next Generation of Target Screening—Quality over Quantity
The subject of target identification and validation has received a lot of attention in the literature in the past year.6,7 This is no surprise as across therapeutic areas, in general, there has been a very low success rate in developing drugs with novel mechanisms (although admittedly, certain areas of oncology, hepatitis C virus, multiple sclerosis, and cystic fibrosis are in the middle of a flush of success).8,9 For several decades, spurred on by the completion of the mapping of the human genome, traditional CNS drug discovery in pharma has employed a systematic approach beginning with high-throughput screening (HTS) of large-scale chemical libraries to find compounds that affected a target of interest. This led to the identification of “lead” compounds and backups of similar chemical series that would then progress to in vivo screening and assessment of “efficacy” in animal models. Although this traditional approach has been a dominating strategy and relatively effective in the identification of the “me too” drugs, this model has proven less successful in identifying novel mechanisms with high probability of clinical success. 10 Not only do new compounds with novel mechanisms of actions have to satisfy statistical improvement relative to placebo, but there must also be a demonstration of improved efficacy relative to the standard-of-care comparators, as well as a reduced propensity for side effects, leading to a level of “differentiation” for any new compounds.
The initial target identification and validation may be a significant contributor to our failure. It is possible that we have been working on targets that are simply not relevant in disease. Very few CNS targets have had compelling supporting data from the very same human diseases they are expected to treat (e.g., genetic or postmortem). Another factor is how we then study those targets, regardless of the strength of the supporting data. Screening an overexpressed target, possibly artificially coupled to a reporter protein of various flavors, in a fibroblast cell line, is about as far away as one can imagine from the target’s native environment, where it may be found in signaling complexes under exquisite regulation, in discrete subcellular locations. Within the native system, you may expect regulatory and compensatory mechanisms reacting to the exposure to compounds. Interestingly, a recent review highlighted the fact that there has been greater success in identifying new drugs from using phenotypic screens. 7 In this case, where one is agnostic of the specific target of interest, the outcome of the screen provides evidence of pathways that may be associated with certain diseases. Of course, it is important to understand the mechanism of any compounds once identified, through de-convoluting the chemical hits, but the phenotypic screen takes the critical step of establishing activity in a native system. 11 This should increase the chances of seeing maintained functional effects as a potential medicine moves through animal and eventually human studies.
Induced Pluripotent Stems Cells—A Possible Component of the Future Drug Discovery Ecosystem
In addition to phenotypic screens, recent technological advances have pointed to the possibilities of screening cells derived from induced pluripotent stem cells (iPS cells). The fundamental discoveries to develop this technology were recently awarded the 2012 Nobel Prize for medicine and have had a huge impact on biomedical research in a very short time. 12 Although this technology is still in its infancy, it enables researchers to obtain cells from a patient, reprogram the cells back into a pluripotent state, and then push them down selected developmental pathways to form neurons or other cell types that are relevant to the patient’s disease. 13 Screening against such cell types is being seen as an important new approach to improve our ability to identify compounds whose effects may translate across assays and survive through development. In addition, it may allow us to have a clearer “line of sight,” particularly at the genome level, from a cell assay, into animal models, and finally into patients with related genomes/risk profiles. Promising examples can already be found across many disease areas, including Alzheimer disease, 14 schizophrenia, 15 Parkinson disease, 16 and diseases on the autism spectrum.17,18 Thus, the use of iPS cells could form part of a platform for future drug discovery projects, allowing for early patient stratification strategies and better selection of relevant animal models ( Fig. 1 ). These are very early days for the technology and its application, though, and robust characterization and utilization of the approach across organizations and emerging precompetitive consortia should determine its real value.
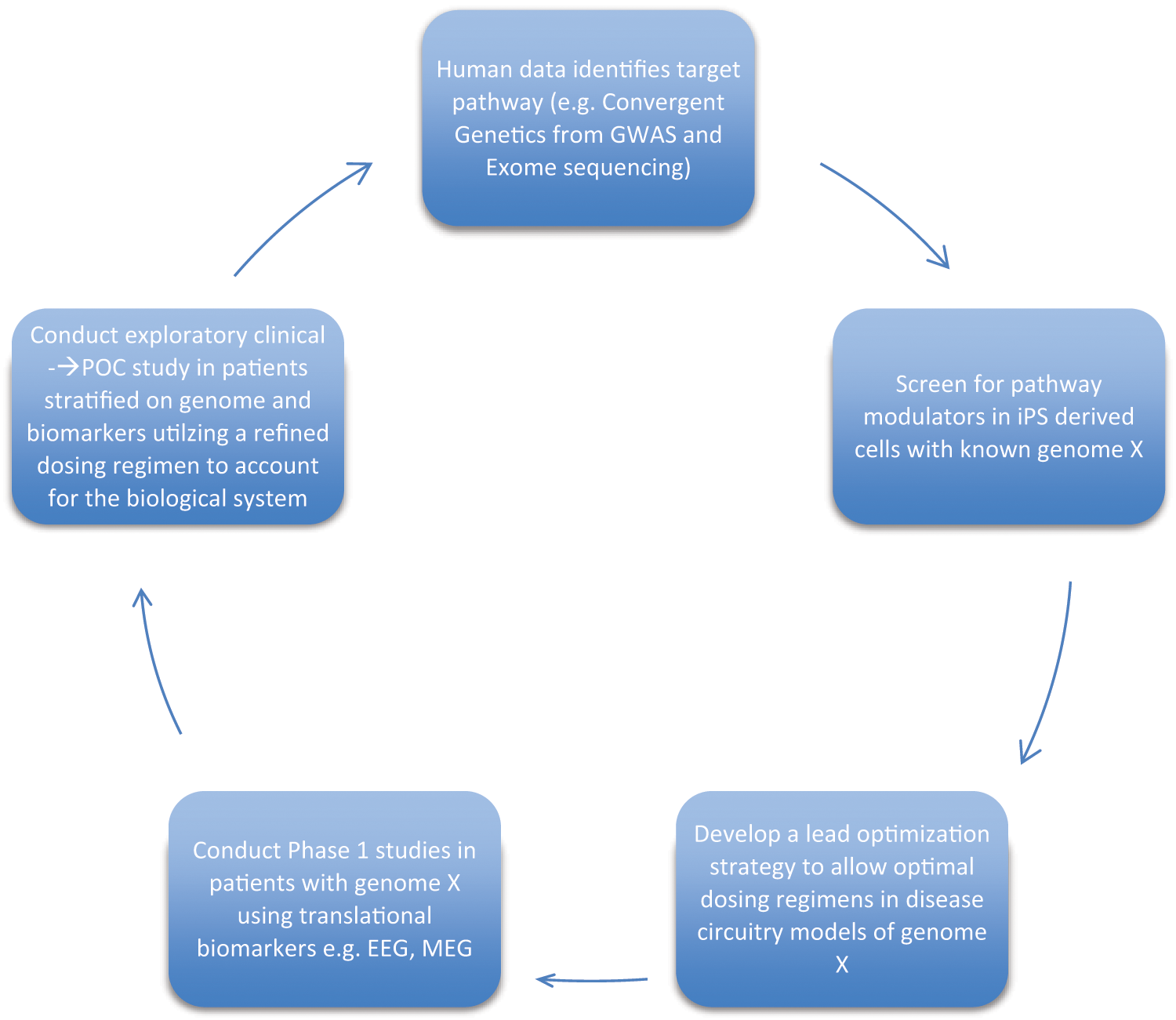
The new cycle of drug discovery and development for CNS therapeutics. The focus on human genetics will guide the initial target identification, validation, and preclinical efficacy strategies. Using emerging integrative electrophysiological and imaging approaches, there will be more confidence in end points at the level of brain circuitry, which should translate into human studies. It is imperative that data from Ph1 and Ph2 studies then feed back into the discovery process. We must learn from our successes and failures and be courageous to not walk away from mechanisms after a single clinical failure. POC, proof of concept; GWAS, genome wide association study; EEG, electroencephalography; iPSC, induced pluripotent stem cell; MEG, magnetoencephalography.
The Preclinical Model Conundrum—The Need for Translatability and Predictive Validity
For several decades, in vivo screening trees have been employed as systematic, tier-by-tier screening approaches for demonstrating in vivo activity of lead compounds and often predesignated for a specific therapeutic area (e.g., antidepressant, Fig. 2A ; antipsychotic, Fig. 2B ). Relative to the in vitro HTS approach used to identify drug candidates, in vivo screening has often been suggested as the bottleneck for drug discovery such that more rapid screening approaches were required.19,20 Thus, the job of the preclinical psychopharmacologist was to select in vivo assays amenable to rapid screening of compounds, albeit at a cost: With the increase in efficiency, there comes a loss of depth in understanding of the underlying biological and physiological processes that result in drug-related behavioral changes. With the increasing number of clinical trial failures in psychiatry, there has been substantial discussion (and fingers pointed) as to whether data generated from in vivo models are predictive or simply misleading. In this respect, it is important to outline a number of obvious criticisms and provide examples for the reader. First, the term efficacy, when used to describe a measure of activity change in a preclinical assay, is misleading and has been loosely used and made interchangeable with activity. Second, it is critical to distinguish the term assay from model, such that an assay should be considered a method of measurement (e.g., immobility in the forced swim test, prepulse inhibition, conditioned avoidance responding), whereas the term model should be understood as a relevant construct of a disease (e.g., disease-relevant mutation, environmental stressor). A third criticism highlights the need to move away from the term predictive validity, a phrase historically used in drug discovery as a designation for an in vivo assay in which a clinically effective class of medicines all show the same behavioral response, such that any compound profiled in the assay that produces a similar behavioral profile would be predicted to be “efficacious” in the clinic. A number of examples of traditional “predictive” preclinical assays can be used to test these criticisms. For the purpose of this review, the forced swim test (FST) will be assessed to represent the predictive antidepressant assays, whereas the conditioned avoidance responding (CAR) assay will be dissected to represent predictive antipsychotic assays. The standard clinically efficacious antidepressant fluoxetine, a selective serotonin reuptake inhibitor (SSRI), produces a reduction in immobility time in the FST following acute administration. Thus, compound X that produces a similar behavioral profile would be predicted to have clinical antidepressant efficacy. 21 Several problems persist with this interpretation. First, with respect to criticism 1, since efficacy in depressed patients is measured by verbal or written responses to a questionnaire (e.g., Hamilton Depression Rating Scale [HAM-D]), the behavioral change induced in the FST in a rodent is a measure of activity in a normal healthy, nondepressed animal (activity not efficacy). With respect to criticism 2 as above, the FST is an assay with a behavioral readout of a change in immobility time (not a model with any construct of depression). With respect to criticism 3, compound X may have a similar pharmacological profile to the standard used for validation in this assay (e.g., monoaminergic-based antidepressant), and therefore the assay is more likely informing us on the level of “predictive pharmacology.” However, this may present concern if novel mechanisms of action evaluated in this assay produce a behavioral profile divergent from the standard reference compound used to validate the assay and provides an example of the limitations of the traditional preclinical screening strategy for antidepressants with novel mechanisms of action. In the CAR assay, all clinically efficacious antipsychotic agents (e.g., haloperidol, olanzapine) produce reductions in avoidance responding in this model. 22 Compound Y, which produces a similar behavioral profile in CAR, would therefore be predicted to have efficacy in patients with schizophrenia. With respect to criticisms 1 and 2, efficacy for antipsychotic agents in clinical patients is often measured using a Positive and Negative Syndrome Scale (PANSS) score, and thus the reduction in avoidance responding produced by compound Y in CAR in otherwise healthy (not psychotic) animals is a measure of behavioral activity in an assay—not efficacy in a model of schizophrenia. Also worth mentioning is that in this behavioral assay, antipsychotics actually reduce an intact behavioral response (avoidance of a perceived threat), which further argues the translatability of this end point. 23 With respect to criticism 3, given that many currently available antipsychotic agents have antidopaminergic mechanisms of action, the predictive pharmacology of this assay would suggest that compound Y may also have a dopaminergic component and not necessarily that it will be antipsychotic.
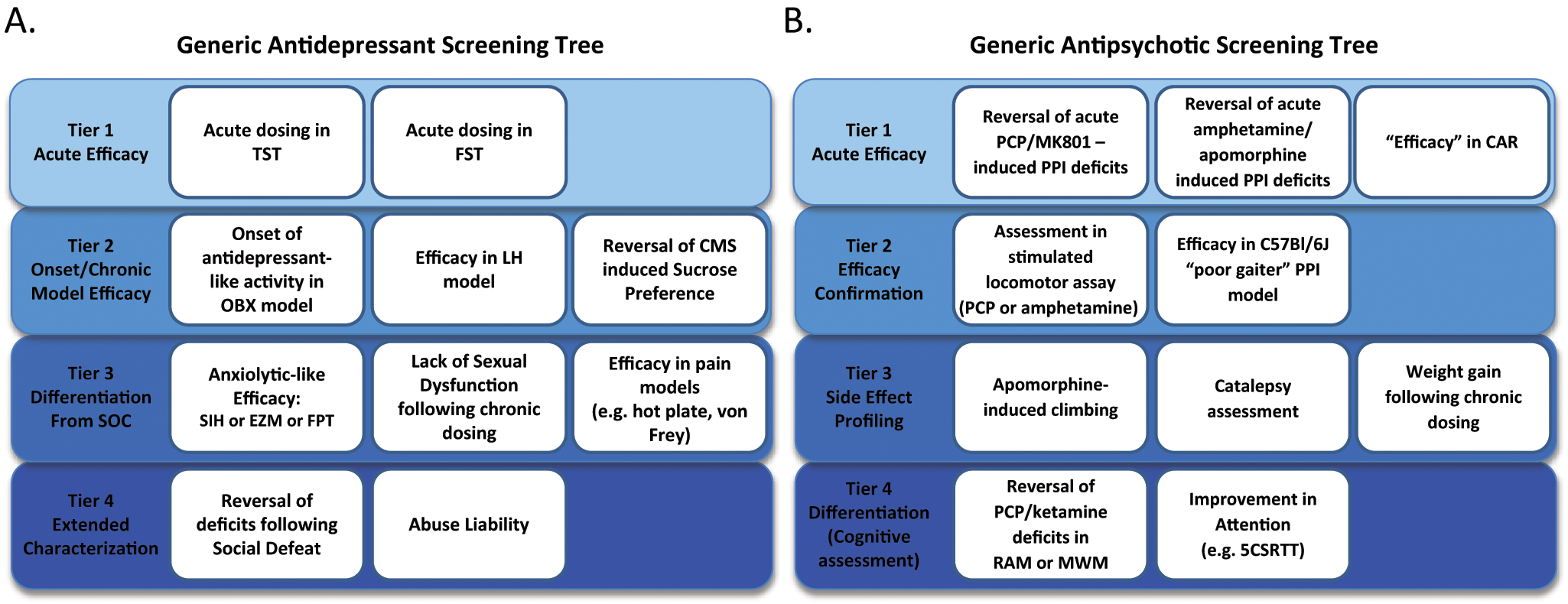
Examples of typical in vivo screening trees traditionally employed to assess preclinical “efficacy” for potential antidepressants (
As highlighted in the recent review of schizophrenia drug discovery by Pratt and colleagues, 24 one of the key factors that preclinical models for psychiatry indications have been lacking is construct validity. Unquestionably, preclinical models that incorporate aspects of the pathophysiology of psychiatric disorders will be more relevant to the human disease. The range of models used in preclinical schizophrenia research has also been recently reviewed by Jones and colleagues. 25 Briefly, disease-relevant pathophysiology or perturbation could be in the form of a genetic mutation that is observed in humans (e.g., 22q11 deletion syndrome, DISC1, or ErbB4; see below), a neurodevelopmental insult such as neonatal administration of phencyclidine (PCP), or in utero exposure to an inflammatory challenge (e.g., polyinosinic:polycytidylic acid, “poly:IC”) or a neurotoxin (e.g., methylazoxymethanol acetate, “MAM E17”). 26
Genetically modified mice have been created that incorporate alterations in some of the most interesting candidate genes in schizophrenia. One example of such a model is the 22q11 syntenic deletion in mice. 27 In humans, this microdeletion accounts for ~1% of schizophrenia cases and increases the risk of schizophrenia ~30-fold in carriers. Multiple mouse models of this deletion differ in the precise genes deleted.28,29 The phenotype of these mice supports their relevance to schizophrenia, with deficits in prepulse inhibition (PPI) and cognitive function. It remains to be seen whether these mice exhibit any behaviors that could be related to negative symptoms in humans. Disrupted in schizophrenia 1 (DISC1) is another example of a genetic mutation associated with schizophrenia. A balanced translocation of this gene was found to be associated with major mental illness in a large Scottish family. 30 Since this discovery, this gene has been the basis of a number of genetically engineered mouse models, from single-nucleotide mutations, to dominant negative transgenics, to in utero RNAi knockdown. The depth of the phenotypic characterization of various Disc 1 mouse models, in terms of behavior, synaptic function, biochemistry, and neurodevelopment, as well as their implications for drug discovery in schizophrenia, has recently been reviewed. 31
An example of a preclinical neurodevelopmental model with potential relevance to schizophrenia is referred to as MAM E17. 32 This model involves the disruption of rat brain development through in utero (E17) treatment with a DNA methylating agent, methylazoxymethanol acetate (MAM). This manipulation causes histopathological changes in adult offspring that are reminiscent of changes observed in schizophrenia, such as reduced hippocampal and prefrontal cortex volume, disruption of corticostriatal circuit physiology, and hallmark deficits in sensorimotor gating and cognitive function. 32
Unfortunately, the current lack of candidate genes identified for major depressive disorder means that there are no promising mutations to recapitulate in engineered preclinical models. Similarly, the depression field has not experienced the same degree of success in developing neurodevelopmental insult models. As a result, it is widely criticized that much of the drug discovery efforts in depression rely on models of behavioral despair in normal rodents (e.g., FST, tail suspension test [TST]), which lack construct validity. We would argue that such models still have utility as assays, when combined with appropriate disease constructs or even more relevant quantitative end points. For example, rather than measuring the effect of a putative antidepressant on immobility time in a normal rodent, this assay, when combined with a “model” that recapitulates an aspect of depression (e.g., cytokine-induced depression), can be used as a pharmacodynamic end point for evaluating potential therapeutic utility of a novel compound. For example, we have recently demonstrated the validity of the interleukin-6 (IL-6)–induced rodent model of depressive behavior. In this paradigm, central administration of recombinant mouse IL-6 produces a depressive-like phenotype, where SSRIs fail to adequately produce a behavioral response, but the behavioral phenotype (increased immobility time in the TST relative to wild-type [WT] can be normalized by administration of an anti–IL-6 antibody. 33 The distinguishing feature of this model-assay combination is that a priori, it would not have been expected to show a classical antidepressant-like behavioral effect (reduced immobility) of the anti–IL-6 antibody when administered to normal animals, but importantly, the anti–IL-6 antibody was capable of normalizing an abnormal behavioral phenotype in a disease-relevant model (increased IL-6 in the CNS). It remains to be seen, however, whether novel mechanisms with anti-inflammatory properties would demonstrate activity in this model and provide translational value.
In an interesting twist, clinical data demonstrating efficacy of acute ketamine in treatment-resistant depression are robust, yet the preclinical literature describing the effects of ketamine in antidepressant assays in rodents is quite mixed—some groups demonstrate acute and sustained efficacy of low doses of ketamine in assays such as FST and TST in normal healthy animals, whereas others only see efficacy after higher doses or only acutely.34–36 Based on our previous criticisms above, it would be of value to pursue the preclinical ketamine studies in cohorts with a disease- relevant construct, perhaps in a transgenic mutant with an overactivated glutamate system, consistent with the hyperactive glutamate levels reported in depressed patients.
Modeling the Diseased Brain
Taking some key points from the previous sections and moving the discussion along, it is clear that the past decades of CNS drug discovery have failed to appreciate that the “brain biology” of a patient with a CNS disorder is likely to be very different from that of a healthy volunteer and truly distinct from that of an inbred rodent. A good example of this is the recent trial of the schizophrenia drug pomaglumetad methionil (LY2140023), a metabotropic glutamate receptor type 2/3 agonist, which had shown promise as a novel antipsychotic. 37 Unfortunately, more extensive phase II and III trials have not met their primary end points (see https://investor.lilly.com/releasedetail.cfm?ReleaseID=703018). 38 A recent publication has suggested that prior exposure to antipsychotics may have downregulated levels of the target of this drug (mGluR2) through regulation of histone deacetylase 2 (HDAC2). 39 It would be expected that patients in these negative trials would have had many years of exposure to antipsychotics, and as such, their “brain biology” is likely to be very different from that seen in a drug-naive patient or a healthy volunteer. This further demonstrates the need to not only improve our understanding of the patient population but also to refine our preclinical modeling to recapitulate the biology of both the disease and a drug-experienced brain, such that the assessment of a novel compound of interest might have improved translatability.
Refinement of Dosing Paradigms
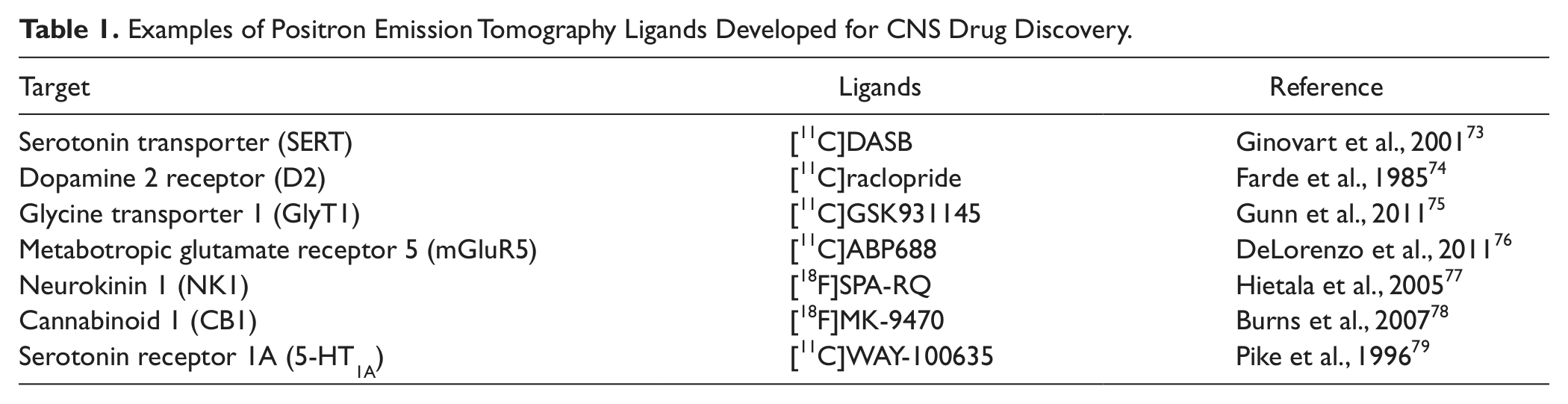
Dose selection is a critical part of the drug discovery process, whether it is in preclinical species or man. We will not discuss this area in detail but focus on one area, which we believe could have far-reaching impact. In most drug discovery units, upon identification of a “compound of interest” (a compound that has appropriate physicochemical, biological, and pharmacokinetic characteristics for preclinical characterization), it is dosed in preclinical studies at a wide range of doses. More recently, greater efforts have been made to estimate the level of target engagement after a given dose of compound. This can be done by measuring free, unbound drug concentration in the brain relative to the affinity (Ki) of the drug for the target. For targets where radioligands exist, measurement of ex vivo or, preferably, in vivo binding in preclinical species is a very powerful technique (see review by Grimwood and Hartig 40 ). Where suitable positron emission tomography (PET) ligands have been developed, the measurement of target occupancy can be translated from rodent binding to nonhuman primate (NHP) and human PET studies. The development of PET ligands is a very high priority for drug discovery, as can be seen by the number of targets for which highly selective PET ligands have been characterized in humans ( Table 1 ; for comprehensive review, see Jones and Rabiner 41 ). Another method that can be used to understand the relationship between brain exposure of a compound and target modulation is in vivo microdialysis. This method has long been used to measure changes in neurotransmitter levels in response to drugs in preclinical species, but it can be taken a step further to monitor the pharmacokinetic (i.e., drug levels) and pharmacodynamic (i.e., neurotransmitter levels) measures simultaneously. 42 Once the plasma exposure required to achieve the desired target occupancy or measurable biomarker of target modulation associated with “efficacy” is known, acute and chronic safety and toxicology studies can be performed to establish the therapeutic window of the drug. The importance of demonstrating sufficient levels of drug exposure, target binding, and functional pharmacological activity—described as “the 3 pillars of Survival”—has been highlighted in a recent perspective. 43 The lack of chronic dosing studies in most industry drug discovery programs, especially in psychiatry, has been a real gap. More demanding, resource-intensive experiments taking advantage of contemporary quantitative pharmacokinetic/ pharmacodynamic (PK/PD) modeling and modern dosing tools such as programmable mini-pumps that can deliver drugs at variable rates to mimic oral dosing PK in humans will be required. Not only does the requisite exposure for efficacy in the desired preclinical model need to be considered, but so does the biological response of the brain to the drug. Compensatory mechanisms in the brain in response to chronic exposure to a drug have rarely been studied adequately in drug discovery. This is not simply about “tachyphylaxis,” a phenomenon of rapid tolerance seen for a range of different drugs where, for example, chronic activation of a receptor leads to downregulation of the receptor and loss of efficacy. Rather, the interest here is in the rebound plasticity shown at the individual cell level and manifesting at the circuit and behavioral level, where different pathways are modified as a response to the drugs’ effects. In many ways, this can be considered analogous to drug resistance seen in cancer cells, but instead of new mutations and pathway activation driving cell plasticity and cell proliferation, we are dealing with the intrinsic plasticity of neurons to change their activity levels. There are a number of ways to overcome this “neuronal resistance.” Modification of the frequency and duration of drug exposure could provide a simple solution. For example, instead of covering the target receptor or enzyme with a certain concentration of drug 24/7, we might want to have a certain level of exposure for a limited period of time. Clearly, we need to ensure that we retain efficacy, but there is precedence for this approach in CNS disorders. One emerging example is in the use of antipsychotics, where intermittent and not continuous dosing is shown to maintain efficacy and also potentially reduce some of the side effects with this medication class. 44 This review by Remington and Kapur 44 succinctly describes the clinical rationale for wanting to reduce dosing frequency of antipsychotics and highlights the clinical data showing that reducing dosing frequency through so-called dosing holidays produces a maintenance of therapeutic effect. In addition, they describe preclinical data that begin revealing the mechanisms behind the differences observed between continuous versus intermittent exposure or “bathing” versus “pinging.” 45 Another different but intriguing example was provided recently in a nonhuman primate cognition model. Dosing with a nicotinic α-7 agonist compound led to improvements in working memory, which incredibly lasted for weeks after the compound exposure. 46 However, in this study, the compound showed efficacy only at very low exposures, while it impaired performance at higher doses. 46 Notably, within the past year, there have been positive clinical signals reported for improving cognitive deficits in schizophrenia with this mechanism, and intriguingly at lower doses, as exemplified by EVP-6124. 47 This gets to another key failure of our (in)ability to translate animal data into the clinic. In so many instances, experimental drugs have been dosed to patients at the highest tolerated dose with an expectation that this would lead to greater efficacy, but this is just not the reality in CNS drug discovery. It is critical that we appreciate these PK/PD observations as we look to translate into patients, particularly because we have clear evidence now that these effects are also seen in humans. This is perhaps best exemplified by dopamine-dependent regulation of cognitive behaviors and “cortical tuning” in the prefrontal cortex. 48 The “inverted-U”–shaped dose response in this system is well accepted, and we have to take notice of this biology in drug discovery. Overall, there is promise that a more refined analysis of dosing regimens could lead to a greater chance of seeing a clinical signal, a reduction in side effects, and, in the long-term, a more sustained period of efficacy. It will be important to integrate this thinking into preclinical assay development.
Examples of Positron Emission Tomography Ligands Developed for CNS Drug Discovery.
The Journal of Replication and Negative Results
We have already scrutinized the lack of ability of the traditional preclinical assays to predict success in the clinic, but it is also important to address other areas in the drug discovery process that have a need for resurgence. In particular, there have been some damning review articles, including a nice report from Bayer scientists, describing how data sets supporting new targets have not been reproduced by independent researchers.49,50 Furthermore, variability in methodologies across laboratories, as well as lack of randomization and blinding in preclinical studies, likely contributes to poor reproducibility. 51 These important aspects are being addressed by precompetitive consortia such as the Innovative Medicines Initiative (IMI) and, specifically, the NEWMEDS project (Novel Methods leading to New Medications in Depression and Schizophrenia), where development of consistent paradigms is a priority (http://www.imi.europa.eu/content/newmeds). With research groups purportedly failing to reproduce a large proportion of published findings (some estimates as high as 50%), it clearly sets up for significant attrition in later development. 50 We are certainly not the first to suggest that publishing negative data should be a welcomed opportunity, but we also encourage publication of results from experiments that fail to replicate published findings. 52
Future Approaches—Bedside to Bench and Back Again
Behavioral pharmacology remains a critical part of CNS drug development, but in its current application, its limitations are clear. Tasks that can be learned efficiently by laboratory rodents tend to be too simplistic to disentangle the host of cognitive processes that all interact to create behavioral performance (e.g., working memory, attention, executive control, motivation). Yet the need for new and more effective CNS medications is enormous and continuing to grow, and advances in both clinical and preclinical research are charting a new path forward for CNS drug discovery. Three key areas of need for moving forward toward a future viable and sustainable CNS drug discovery program are suggested: (1) improved preclinical disease-relevant modeling, (2) gaining a mechanistic understanding of CNS functions and potential drug target effects at the level of neural circuitry, and (3) improved translational measures and back-translation of learnings from both clinical failures and successes.
Preclinical end points should move away from traditional generic behavioral screening trees in normal, otherwise healthy animal subjects and employ a combination of disease-relevant approaches ( Fig. 3 ). As mentioned earlier, these should include animal models incorporating underlying genetics of human disease (e.g.; 22q11, neuroligin, DISC1 mutations), pharmacological perturbation relevant to target biology (specifically not employing acute, transient pharmacological disruption but rather chronic or neonatal treatment [e.g., neonatal PCP]), or environmental manipulations that encompass a neurodevelopmental perturbation (e.g., MAM, poly IC).
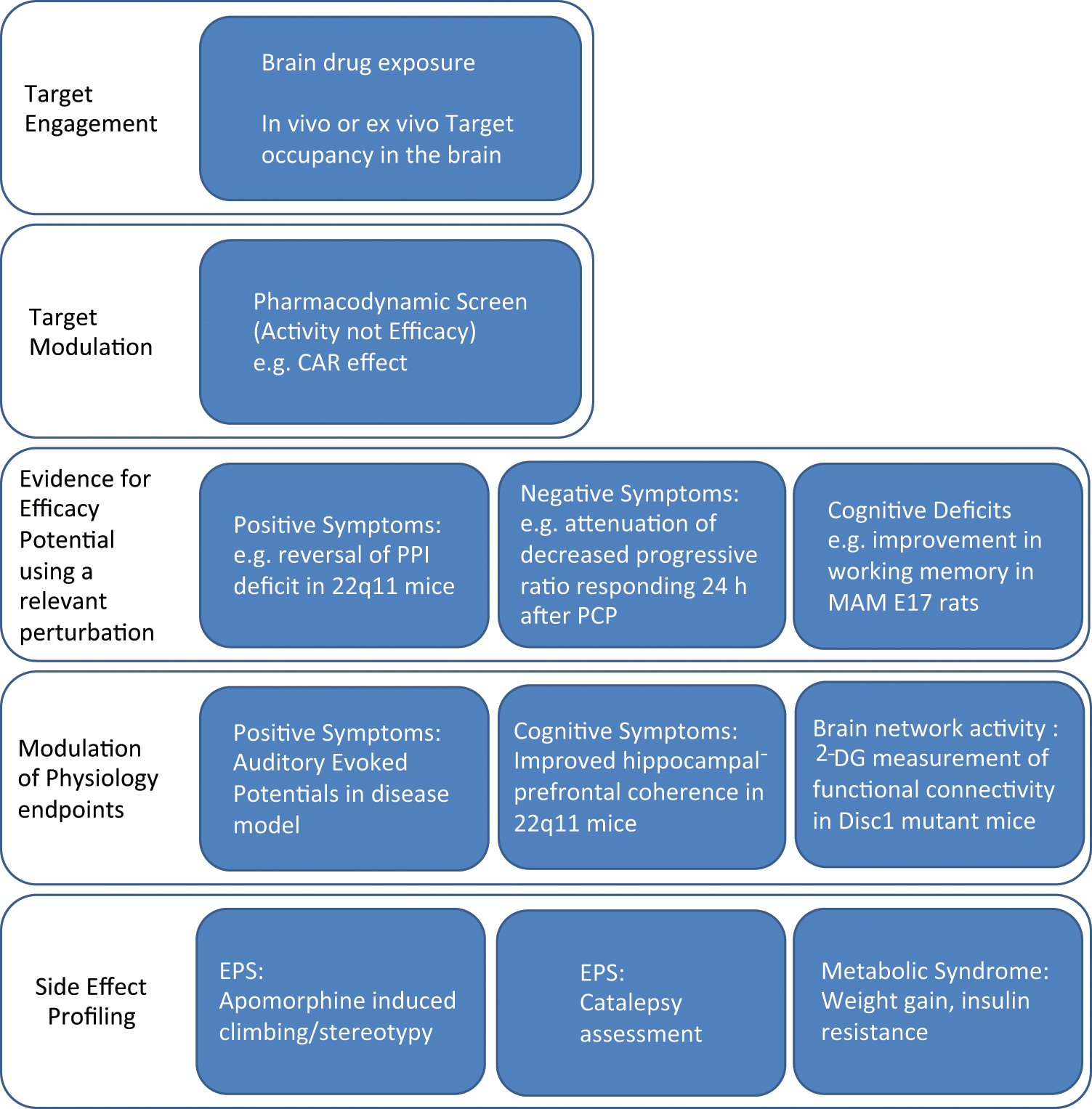
A proposed screening tree for evaluation of novel putative antipsychotic agents incorporating disease-relevant models. Characterization of lead compounds begins with confirming target engagement, either through measurement of compound exposure in the brain or, preferably, if suitable radioligands exist, through measuring target occupancy. At doses that occupy the target, a compound would have to demonstrate a pharmacodynamic effect, that is, a behavioral effect that does not have to be related to efficacy (e.g., conditioned avoidance responding, CAR). Efficacy should then be demonstrated in a disease-relevant model or perturbed system (genetic, pharmacological, or neurodevelopmental). To demonstrate differentiation from standard antipsychotics, a lead compound would have to demonstrate efficacy in ≥2 assay-model combinations (e.g., prepulse inhibition, PPI). Compounds meeting this criterion would proceed to more involved physiological assessment (e.g., in vivo electrophysiological recordings or 2-deoxyglucose [2-DG] imaging) to probe effects on circuit function and to provide translatable end points that would form part of the early clinical plan. As with the traditional paradigm, a novel compound would still need to demonstrate limited propensity for side effects associated with existing therapeutics (extrapyramidal [EPS] and metabolic). Collectively, information from all stages of this screening paradigm would be used to determine the relationships between exposure, target occupancy, and behavioral and physiological end points to identify the range of drug exposure concentrations to target in humans. MAM E17, methylazoxymethanol acetate; PCP, phencyclidine.
Moving forward, there will continue to be a push for translatable end points in psychiatry, as well as information back-translated from the clinic that informs the discovery scientists. One of the best-described efforts for identification of translatable end points is the 5-choice serial reaction time task (5CSRTT), an assay developed to be a rodent analogue of the human continuous performance task (CPT). 53 Unfortunately, despite all these efforts, while there is evidence of the CPT being used in small investigator-initiated clinical studies, 54 utilization of this test by the pharmaceutical industry in the clinical development plans for novel mechanisms thought to have effects on attention is not apparent. PPI, a measure of sensory gaiting, is another such example of an end point reported to be measurable in humans with a defined circuitry that is conserved across species and can be tested using similar protocols from human to rodent. Although there are numerous publications describing disruptions in PPI in a range of clinical disorders (e.g., schizophrenia, obsessive-compulsive disorder, Tourette syndrome; for review, see Braff et al. 55 ), there remain little data on the pharmacological reversal of such deficits and no obvious reports of this end point being measured as part of clinical development plans. This may be due to initial reports that investigated the presence or absence of PPI deficits in schizophrenia patients treated with typical or atypical antipsychotics, yielding mixed results. Some studies reported that deficits in PPI persisted in patients on typical antipsychotics, whereas others indicated that patients treated with atypical agents either did or did not have deficits in PPI. 55 One important factor highlighted in the review by Braff and colleagues 55 was the variability in experimental paradigms across different studies. This highlights the value of consistent experimental design, both in preclinical and clinical settings, to get an understanding of the degree of variability and reproducibility of a given assay.
Boldly Going Beyond—The Possibilities of Circuits
In recent years, efforts to develop new therapies for CNS disorders have increasingly recognized that abnormal activity in specific neural circuits underlies many of the disease symptoms. This has affected the strategy for developing new treatments in two major ways: First, neurosurgical approaches using focused brain stimulation to normalize circuit activity have shown promise in a growing list of CNS disorders that now includes Parkinson disease, dystonia, epilepsy, Tourette syndrome, and, more recently, obsessive-compulsive disorder and treatment-resistant depression. 56 Second, efforts to develop new therapeutics are increasingly using measures and manipulations of neural circuit activity to understand disease symptoms and to evaluate drug efficacy. As is often the case, technological innovations have played an essential role in this strategic shift. In particular, electrophysiological recordings in awake, behaving subjects; optogenetic manipulation of neural circuits; biosensor measures of tissue oxygen levels; and advanced in vivo optical imaging have opened new horizons for CNS drug discovery.
Recent drug development efforts are emphasizing translational electrophysiology measures such as electroencephalography (EEG) and magnetoencephalography (MEG). Although not able to resolve the activity of individual neurons, EEG and MEG offer noninvasive and reasonably cost-effective ways to measure oscillatory activity among neural populations, and there are now many studies linking neural oscillations to both normal brain function and disease.57–59 Abnormal neural circuit activity patterns have now been identified in a number of CNS disorders, including Parkinson disease, schizophrenia, autism, major depression, Alzheimer disease, and Huntington disease.60–64
For example, patients with schizophrenia appear to have abnormal neural oscillations, specifically a relative reduction in oscillatory activity that is believed to be essential for normal brain function. Oscillations in the gamma frequency range (30–100 Hz) are associated with cortical activation65,66 and are believed to represent a fundamental aspect of cortical information processing. 57 Compared with healthy control subjects, patients with schizophrenia show reduced gamma oscillations in response to external stimuli and during cognitive tasks that normally evoke gamma activity.67,68 This likely involves the significant abnormalities observed in a specific population of cortical interneurons that are critical for the generation of cortical gamma oscillations. 69 Thus, although the abnormal oscillatory activity in patients with schizophrenia is quite different from that in patients with Parkinson disease, there is substantial evidence to indicate that abnormal neural circuit activity patterns are critical aspects of both disorders and should be incorporated into future drug development strategies for treating these patients.
In vivo electrophysiology in behaving animals is perhaps the most direct approach to measuring the neural circuit activity that underlies behavior. Depending on the specific question at hand, one can measure the intracellular membrane potential of individual neurons, the extracellular spiking activity of tens to hundreds of neurons, the local field potential generated by synaptic and intrinsic membrane currents in thousands of neurons, or EEG potentials reflecting the activity of much larger neural populations. 70 Often considered tools of basic science research due to their low throughput, measures such as these are increasingly being incorporated into drug discovery programs because of the industry’s increased focus on neural circuit activity and facilitated by commercial efforts to reduce the difficulty of the technique. Although recording the spiking activity of tens or hundreds of neurons in multiple brain regions of rodents performing tasks remains a slow and arduous undertaking, the potential value of this methodology to drug development for psychiatric disorders is important. Although it may never be possible to accurately assess conditions such as depression and psychosis in rodents, it may well be possible to use rodent models to identify and treat the underlying neural circuit abnormalities that cause the deleterious symptoms of these conditions.
Although electrophysiological recording methods enable researchers to measure neural circuit activity at all levels from large neural populations down to single neurons, the ability to control circuit activity with electrophysiology, thereby directly testing hypotheses about its broader significance, is much more limited. It has long been possible to deliver local electrical current to stimulate the neural tissue, either through the recording electrode or through a second electrode placed nearby, but this method suffers from two major disadvantages: (1) Current injection can easily activate neurons, but it is much more difficult to inhibit a neural circuit using electrical stimulation, and (2) any axons that pass near to the site of stimulation can be excited, regardless of whether they are part of the intended neural circuit. Optogenetics, the functional control of specific neurons using a combination of genetically encoded ion channels and light, has provided a long-awaited solution to this problem. Researchers working with animal models of disease can now simultaneously manipulate and measure the activity of specific neural circuits, and this can be done in awake animals as they perform behavioral tasks. By combining optogenetics and electrophysiology with candidate molecules and/or improved models of disease, it is becoming possible to reproduce abnormal, disease-relevant circuit activity based on clinical electrophysiology, assess the behavioral impacts of that abnormal activity, and then try to correct the circuit with specific available compounds. In this way, optogenetic methods may facilitate the development of new preclinical approaches for modeling human circuit disorders, in addition to the widely appreciated advantages for basic science research these new tools offer.
Electrophysiological methods are optimal for very rapid signals such as neural oscillations and spiking activity but provide only limited spatial information and are not suitable for noninvasively measuring the activity of deep brain structures. Functional imaging, by contrast, can provide a spatiotemporal map of relative activity throughout the brain in a noninvasive manner, making it the method of choice for most clinical studies. Unfortunately, the cost of functional imaging and the difficulty of combining it with behavior in laboratory animals have limited the extent to which functional imaging data can translate from humans to preclinical research animals. A partial solution that has recently emerged is in neurochemistry probes that allow for the real-time detection of tissue oxygen levels in behaving animals. These oxygen biosensors can be surgically implanted in lab animals using similar procedures as for in vivo electrophysiology, enabling researchers to monitor changes in local oxygen consumption in freely behaving animals. Although the spatial information is limited to the region immediately surrounding the biosensor probe, a disadvantage compared with functional imaging, oxygen biosensors provide a signal that reflects the activity of neurons in the region 71 and is believed to be analogous to the functional magnetic resonance imaging (fMRI) blood oxygenation-level dependent (BOLD) signal. 72 Because biosensors can be combined with conventional electrophysiological recordings, this technology not only provides a better means of incorporating clinical imaging observations into preclinical research programs but also promises to help clarify the relationship between electrophysiological measures and the fMRI BOLD signal, enabling preclinical electrophysiology data to be more directly applied to human clinical studies of neural circuit activity.
Implementation of an Integrative Approach
In this review, we have addressed some critical issues within the CNS drug discovery process that need fixing, and we have laid out some basic strategies for achieving success in that regard. Many of the ideas we have discussed are actively being considered across the industry and in the broader research community. For example, the National Institute of Mental Health (NIMH) has recently established the Research Domain Criteria project (RDoC; http://www.nimh.nih.gov/research-funding/rdoc/index.shtml) with an effort to improve the translation of preclinical data into clinical success. The RDoC recognizes that past efforts for CNS drug development have been too strictly guided by the Diagnostic and Statistical Manual of Mental Disorders (DSM) criteria of clinical disease diagnosis. The major psychiatric diseases are heterogeneous collections of disorders that lie on a spectrum, with many shared symptoms, many shared risk factors, and somewhat arbitrary divisions between them. From this perspective, it does not make sense for preclinical scientists to spend their time trying to develop animal models of entire psychiatric conditions such as schizophrenia; instead, animal model development should focus on recapitulating abnormalities at the genetic, cellular, synaptic, and neural circuit levels that underlie specific impairments in broad domains of brain function. A focus on domains of function (e.g., working memory, motivation, attention) not only simplifies the task of developing animal models but also helps researchers to select the relevant brain circuits, behavioral assays, and efficacy measures that dictate the key experiments to be carried out. Thus, the future integrated development strategy for in vivo screening might follow these steps: (1) Identify a relevant domain of function that is impaired in psychiatric patients, significantly affects their quality of life, and is not adequately treated by existing medications; (2) focus on the brain circuitry that underpins this domain of function; (3) develop an animal model using genetics and/or pharmacological insults that produce a disease-relevant dysfunction in this circuitry; (4) validate the animal model by demonstrating an impairment in the chosen domain of function; (5) select efficacy measures based on integrated measures of circuit activity during the translational behavioral assays that probe this domain of function; (6) screen compounds in this integrated animal model/assay combination for acute efficacy; (7) chronically dose the lead compounds and optimize the dosing schedule and chemical matter based on observed compensatory changes; and, finally, (8) take lead compounds into the clinic, carefully stratifying the patient population based on impairments in the selected domain of function and any known genetic factors that link to the target.
In conclusion, the vision of the future drug discovery project discussed herein and illustrated in Figure 3 is one where a project is initially started based on robust data from patient samples, followed up by elegant screening in iPS cells or similarly complex systems. Once a compound has been discovered, it will be characterized in integrated neurophysiological/behavioral paradigms to understand circuits. The end points measured will be already suggestive of what end points we will measure in Phase I in patients or volunteers with stratified genomes. Doses selected in man will cover a range of paradigms to ensure we find the sweet spot for efficacy, safety, and biology. None of this is going to be easy, and it will certainly slow down the discovery process, but it will have an eventual return in time savings, innovative clinical trial design, and ultimately clinical success and return on investment in the later development process.
Footnotes
Declaration of Conflicting Interests
SJSR, JRE, and ZAH are full-time paid employees of Pfizer. NJB is a full-time paid employee of AstraZeneca.
Funding
SJSR, JRE, and ZAH are full-time paid employees of Pfizer. NJB is a full-time paid employee of AstraZeneca.