
Abstract
A variety of G-protein–coupled receptor (GPCR) screening technologies have successfully partnered a number of GPCRs with their cognate ligands. GPCR-mediated β-arrestin recruitment is now recognized as a distinct intracellular signaling pathway, and ligand-receptor interactions may show a bias toward β-arrestin over classical GPCR signaling pathways. We hypothesized that the failure to identify native ligands for the remaining orphan GPCRs may be a consequence of biased β-arrestin signaling. To investigate this, we assembled 10 500 candidate ligands and screened 82 GPCRs using PathHunter β-arrestin recruitment technology. High-quality screening assays were validated by the inclusion of liganded receptors and the detection and confirmation of these established ligand-receptor pairings. We describe a candidate endogenous orphan GPCR ligand and a number of novel surrogate ligands. However, for the majority of orphan receptors studied, measurement of β-arrestin recruitment did not lead to the identification of cognate ligands from our screening sets. β-Arrestin recruitment represents a robust GPCR screening technology, and ligand-biased signaling is emerging as a therapeutically exploitable feature of GPCR biology. The identification of cognate ligands for the orphan GPCRs and the extent to which receptors may exist to preferentially signal through β-arrestin in response to their native ligand remain to be determined.
Introduction
Sequencing of the human genome has revealed the G-protein–coupled receptor (GPCR) superfamily to be one of the largest human gene families comprising on the order of 750 members. 1 These seven transmembrane proteins are ideally positioned to sense molecules in the extracellular environment and transduce signaling information to orchestrate an appropriate cellular response. The range of stimuli acting upon GPCRs is diverse, including neurotransmitters, peptides, lipids, nucleotides, metabolic intermediates, odorants, and light, highlighting the wide-ranging roles of GPCRs in the coordination of mammalian physiology. The GPCR superfamily is commonly divided into three main classes. The rhodopsin-like or class A GPCRs represent by far the largest class, with more than half its members acting as chemosensory receptors for stimuli originating in the extracellular environment. The remaining nonsensory GPCRs have largely been characterized and respond to endogenously derived ligands. Class A GPCRs may be distinguished from class B and class C receptors on the basis of amino acid sequence. Class B and class C GPCRs display a large extracellular domain and a large bilobed N-terminal Venus flytrap domain, respectively. GPCR subgroups may also be differentiated with respect to the position of the orthosteric ligand-binding site and the chemical nature of the natural ligand. Despite intensive investigation, there remain approximately 75 class A, 30 class B, and 7 class C orphan receptors for which endogenous ligands have not been identified. The extent to which receptors may be considered to be partnered with their naturally occurring ligand is often a topic for debate. In this respect, the International Union of Basic and Clinical Pharmacology Committee on Receptor Nomenclature and Drug Classification (NC-IUPHAR) provides an online database to act as a resource of information on 354 nonchemosensory GPCRs. 2 We have adopted IUPHAR recommendations regarding receptor nomenclature, and we cite this resource for the large repository of original publications describing the cloning, ligand pairing, and pharmacology of GPCRs described in this article. The GPCR superfamily has been targeted for the generation of therapeutically useful drugs, and as such, the orphan GPCRs represent an untapped resource of potentially novel targets for both established and emerging disease areas.
Many investigators have sought to identify natural ligands for the orphan GPCRs using a reverse pharmacology approach, whereby the receptor of interest is screened against libraries of putative endogenous ligands and tissue extracts.3–5 Similarly, libraries of natural products and synthetic compounds have been screened to identify surrogate agonist and antagonist ligands to provide a starting point for the study of receptor pharmacology and physiology. GPCR deorphanization programs have continually incorporated advances in high-throughput screening (HTS) technologies. Typically, GPCR overexpression in a cell-based system has been linked to a classical heterotrimeric G-protein–coupled readout, such as intracellular calcium mobilization or cyclic AMP production. It has become increasingly clear, however, that GPCR signaling is not limited to G-protein coupling. Importantly, the β-arrestin family of proteins, originally described as desensitizers of receptor signaling and mediators of endocytotic recycling, are now recognized as distinct G-protein–independent mediators of cell signaling. 6 Ligands acting at a GPCR may also show varying degrees of bias with respect to the potency and efficacy with which they may activate classical heterotrimeric G-protein and β-arrestin signaling. Furthermore, ligand-receptor interactions may show a complete or “perfect” bias toward the activation of the β-arrestin pathway.7–10 G-protein–independent assay systems are now established in the analysis of GPCR pharmacology, and indeed β-arrestin assay technology has been applied to the identification and confirmation of ligands for a number of recently deorphanized GPCRs. 11 Here we describe work aimed at identifying novel ligand-receptor pairings for the orphan GPCRs based on a potential signaling bias toward the β-arrestin pathway. These experiments were made possible by the use of putative ligand banks established at GlaxoSmithKline (GSK; Stevenage, UK) and the availability of an extensive panel of liganded and orphan GPCRs configured using DiscoveRx (Fremont, CA) PathHunter β-arrestin enzyme fragment complementation (EFC) technology.
Materials and Methods
Candidate Ligand Collection
The ligand collection comprised 10 500 individual samples compiled from a number of distinct compound libraries. It was estimated that there was a 10% to 15% overlap between library sets, which was considered a potential benefit given the inherent lability of some sample types. The collection was based around two proprietary GSK collections of ~3500 naturally occurring bioactive molecules (lipids, peptides, neurotransmitters, orphan molecules, cannabinoids, nucleotides, vitamins, sugars) and was supplemented by the inclusion of the Biomol/Enzo (Farmingdale, NY) cannabinoid, lipid, eicosanoid, and fatty acid sets (~1000 compounds) and the Phoenix (Burlingame, CA) peptide set (~800 compounds).
In addition to candidate endogenous ligands, we included GSK Natural Product–like compounds (~2700 compounds) and synthetic compounds from the National Institutes of Health (NIH) clinically relevant small-molecule set (~450 compounds) and the Sigma (St. Louis, MO) LOPAC small-molecule set (~1280 compounds). This enhanced the overall diversity of the screening set and acted as an indicator of the tractability of the orphan GPCRs as targets using synthetic ligands. The publicly available content of the ligand collection may be accessed via
PathHunter β-Arrestin Cell Lines
The PathHunter β-arrestin cell lines provided by DiscoveRx are listed in
Evaluation of Receptor Expression
Receptor expression and glycosylation status of the ProLink-tagged orphan GPCR were assessed in each cell line by PathHunter EAstern Blot analysis according to the manufacturer’s instructions (DiscoveRx). Briefly, cell lysates containing ProLink-tagged fusion proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a nitrocellulose membrane, and incubated with enzyme acceptor reagent to facilitate complementation of β-galactosidase activity. Chemiluminescent detection of the ProLink tag confirms the expression of the GPCR. Pretreatment of cell lysates with PNGase to cleave N-linked oligosaccharides demonstrated correct protein molecular weight and the glycosylation status of the expressed receptor. A monoclonal antibody was generated against the ProLink sequence in mice immunized with the synthetic peptide CPFASWRNSEEARTDR conjugated to tuberculin purified protein derivative. Splenocytes from a seropositive animal were fused with P3X63/Ag8.653 myeloma cells and culture medium subsequently tested for antipeptide activity by indirect enzyme-linked immunosorbent assay (ELISA) using 96-well plates coated with bovine serum albumin (BSA)–conjugated peptide. A hybridoma was subcloned by limiting dilution and immunoglobulin (IgG2a) purified by protein A chromatography. Cells were prepared for flow cytometry using FIX & PERM reagents (Life Technologies, Paisley, UK). Briefly, cells were permeabilized and labeled with anti-ProLink monoclonal antibody or isotype control for 20 min at room temperature (RT). Cells were washed and stained with an FITC-labeled secondary antibody for 20 min at RT. Cells were washed, resuspended in buffer, and processed for flow cytometric analysis (Becton Dickinson, Oxford, UK).
PathHunter β-Arrestin Recruitment Assays
Prior to screening, cells were removed from normal growth medium and incubated for 24 h in Cell Plating Reagent 1 (DiscoveRx). The cells were recovered using enzyme-free cell dissociation reagent and adjusted to a density of 0.5 million cells per milliliter in Hank’s balanced salt solution (HBSS) and 25 mM HEPES (pH 7.4) containing 0.1% BSA. Immediately prior to use, each well of a 32-plate screening set was adjusted to a final volume of 5 µL in Opti-MEM1 medium. The assay was initiated by the addition of 20 µL cell suspension (10 000 cells) to each well of a 32-plate screening set. The plates were incubated at 37 °C for 90 min to facilitate β-arrestin recruitment. The plates were equilibrated to RT for 30 min prior to the addition of 10 µL PathHunter detection reagent (DiscoveRx). The detection reaction was incubated for 1 h at RT in the dark and chemiluminescence measured on a Wallac Viewlux microplate imager (PerkinElmer, Beaconsfield, UK).
Intracellular Signaling Assays
Calcium mobilization in PathHunter cell lines was monitored on a Flex Station (Molecular Devices, Winnersh, UK). Cells were seeded in complete growth medium as 10 000 cells per 384-well in black, clear-bottom microplates and incubated overnight. Cells were loaded with Fluo-4 Direct Calcium assay reagent in the presence of probenecid (5 mM) for 1 h at 37 °C (Life Technologies). For CHO cells stably expressing N-terminally HA-tagged human GPR84 in combination with chimeric Gqi5, cells were loaded with Fluo-4AM (2 µM) in the presence of Brilliant Black (0.25 mM) (Molecular Devices) and 2.5 mM probenecid for 1.5 h. Fluorescence was monitored using a FLIPR (Molecular Devices). Cyclic AMP levels were measured in cells incubated with 25 µM forskolin using a HitHunter cyclic AMP assay according to the manufacturer’s instructions (DiscoveRx).
Yeast Gα13 Assay
Saccharomyces cerevisiae expressing a chimeric version of Gpa1, in which the last five amino acids of the yeast G-protein were replaced by the C-terminal five amino acids from human Gα13, were transformed with human GPR35. Receptor coupling through Gα13 promoted cell growth via the yeast pheromone receptor pathway and was quantified fluorometrically. 12
Data Analysis
In the absence of positive control ligands for each orphan GPCR, assay quality was assessed with reference to individual plate means and assay coefficients of variation (CVs). The mean assay CV across 82 GPCRs screened was 12.1% ± 5.2%. Spotfire (TIBCO, Somerville, MA) analysis was used to identify data points significantly greater than three times the standard deviation (SD) of the assay mean to generate a list of potential candidate hit ligands for each receptor, which were then ranked according to the fold-increase in β-arrestin activity. Receptor selectivity was assessed by reference to somatostatin receptor 2 (SSTR2) and by internal reference to groups of orphan GPCRs during ligand confirmation by EC50 determination. Selected candidate hits identified for a group of approximately 12 GPCRs were repurchased and processed for confirmatory dose-response studies against each of the 12 GPCRs. In this manner, batch analysis of candidate hits and receptors afforded a degree of internal control in the de-selection of nonspecific and recurrently active ligands and the selectivity of candidate ligand-receptor interactions. Curves were fitted by nonlinear regression analysis in GraphPad Prism (GraphPad Software, La Jolla, CA).
Results and Discussion
Assay Validation and Screening for Ligand-Induced β-Arrestin Recruitment
Screening quality and performance may often be assessed by the inclusion of known activators or inhibitors of the target of interest to serve as controls on each assay plate. By their very nature, orphan GPCRs lack such control reagents, presenting an issue in terms of assay optimization and quality control. Throughout this program, we incorporated measures to demonstrate that, if not formally optimized, each GPCR assay was appropriately configured and that assay functionality could be demonstrated. Receptor expression in each PathHunter β-arrestin cell line was detected using EAstern blotting technology (DiscoveRx) and confirmed immediately prior to commencing each screen by flow cytometric staining using a monoclonal antibody raised against the ProLink tag sequence integral to each GPCR. A key feature of PathHunter technology is that the EFC signal is specifically derived from the interaction of the ProLink-tagged GPCR of interest with the enzyme acceptor–tagged β-arrestin. The coexpression of these two components was found to generate a basal level of EFC, representing constitutive GPCR–β-arrestin recruitment in the absence of applied ligand and was not seen with either component alone. This basal activity was indicative of a functional interaction between receptor and arrestin and served as the background or low assay control.
The steps undertaken to validate the use of each PathHunter β-arrestin cell line prior to screening are illustrated for the orphan receptor GPR35 (
Identification of Novel Putative GPR35 Agonists
Novel surrogate ligands promoting GPR35 β-arrestin recruitment were also identified (
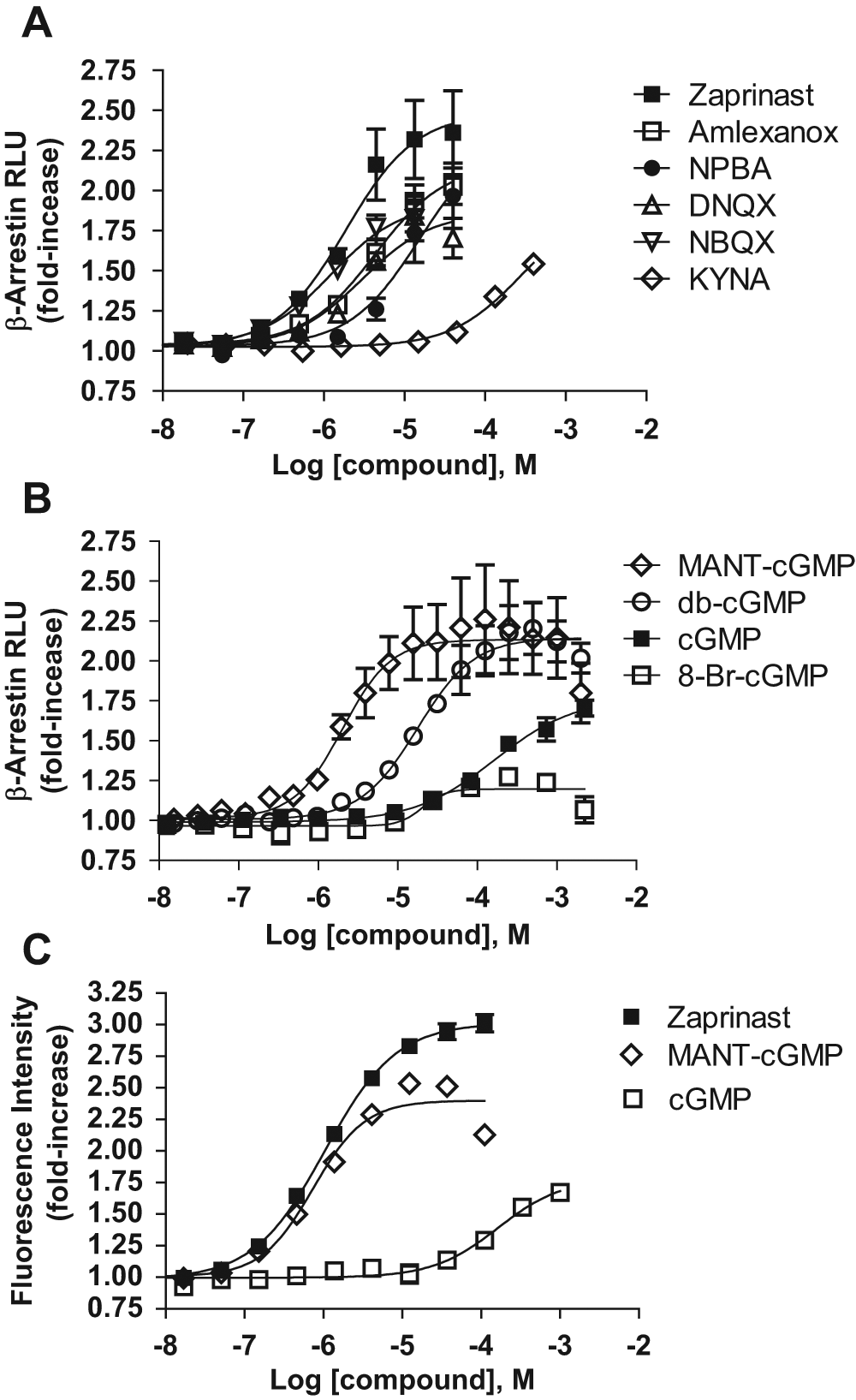
Dose-response curves for PathHunter β-arrestin recruitment to GPR35 by agonists identified in
As noted above, the surrogate GPR35 ligand and PDE inhibitor, zaprinast, is a structural analogue of guanosine-3′,5′-cyclic monophosphate (cGMP). It was therefore of note that we identified the cell-permeant cGMP analogues dibutyryl cGMP, 2′-O-(N-methylanthraniloyl)-cGMP (MANT-cGMP), and 2′-O-anthraniloyl-cGMP also to be surrogate ligands at GPR35 (
We further highlighted the robustness of the screening rocess through the identification of a number of other well-established ligand-receptor pairings, confirming that these GPCRs are indeed likely to be matched with their cognate ligands (
Table 1
and
Confirmation of Ligand-GPCR Pairing by PathHunter β-Arrestin Recruitment.
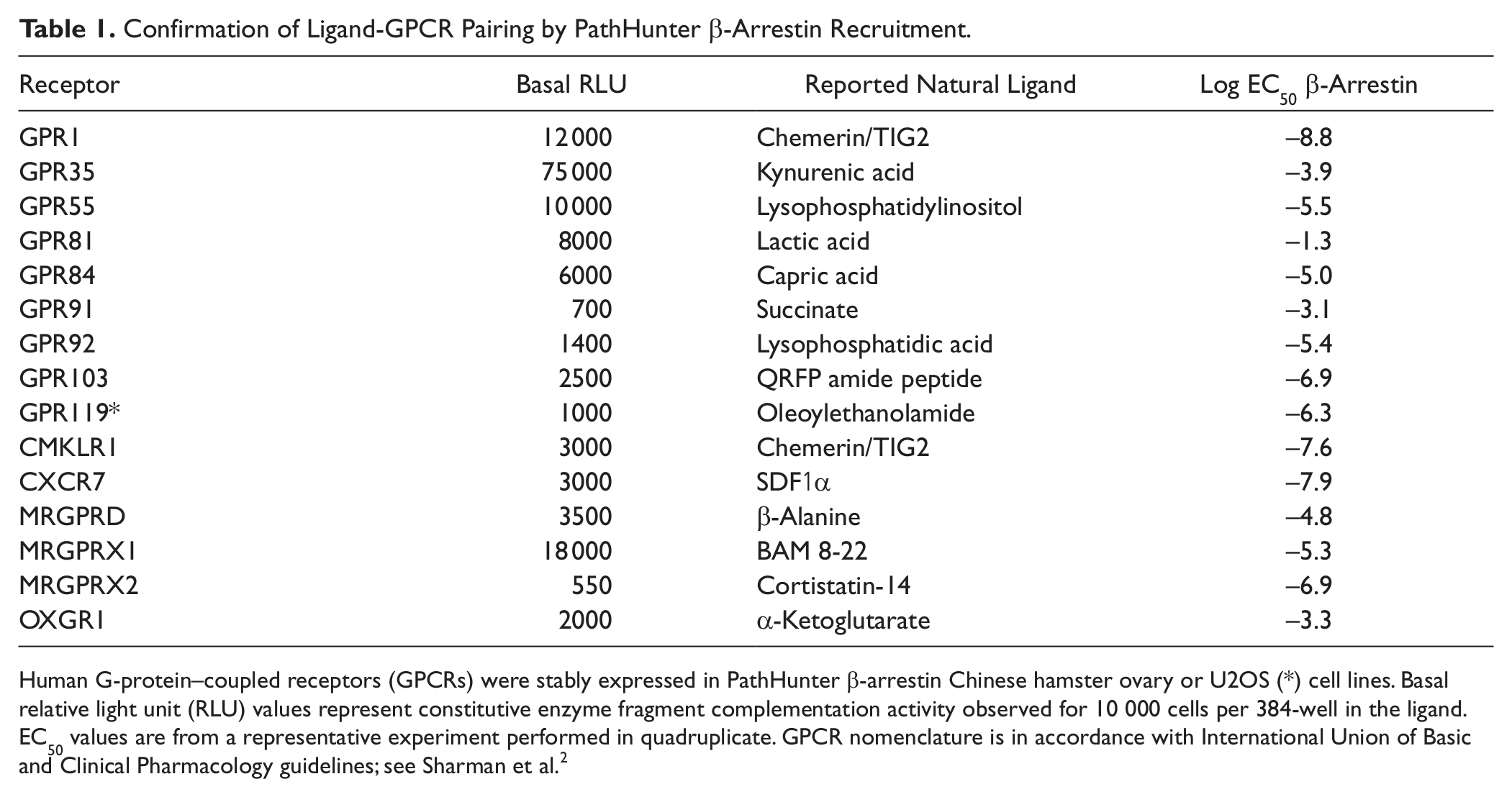
Human G-protein–coupled receptors (GPCRs) were stably expressed in PathHunter β-arrestin Chinese hamster ovary or U2OS (*) cell lines. Basal relative light unit (RLU) values represent constitutive enzyme fragment complementation activity observed for 10 000 cells per 384-well in the ligand. EC50 values are from a representative experiment performed in quadruplicate. GPCR nomenclature is in accordance with International Union of Basic and Clinical Pharmacology guidelines; see Sharman et al. 2
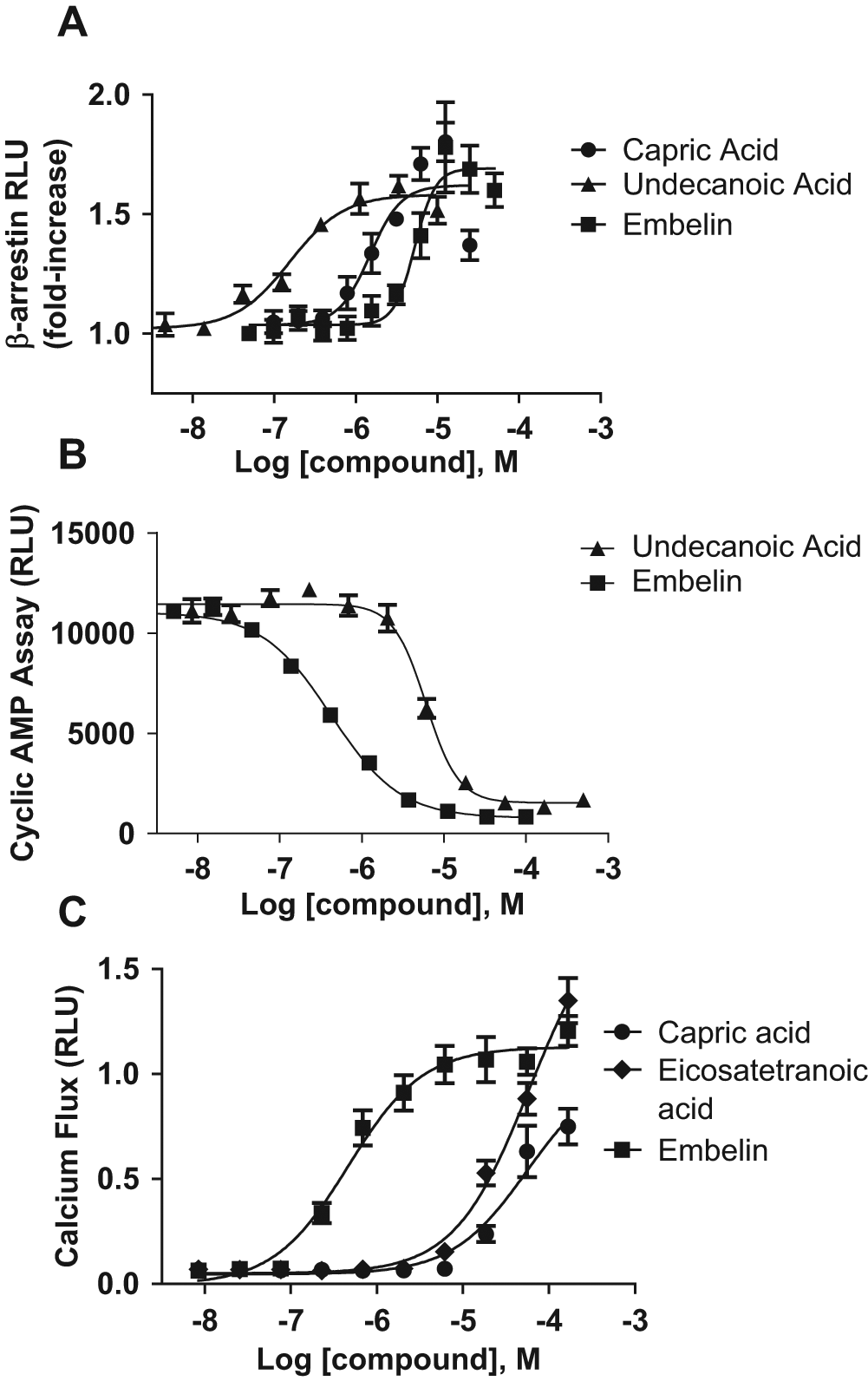
Activation of GPR84 by fatty acids. Medium-chain length fatty acids and the surrogate agonist embelin were tested for their ability to agonize GPR84, measuring activation of (
Ligand-GPCR Pairings That Were Not Confirmed by PathHunter β-Arrestin Recruitment.
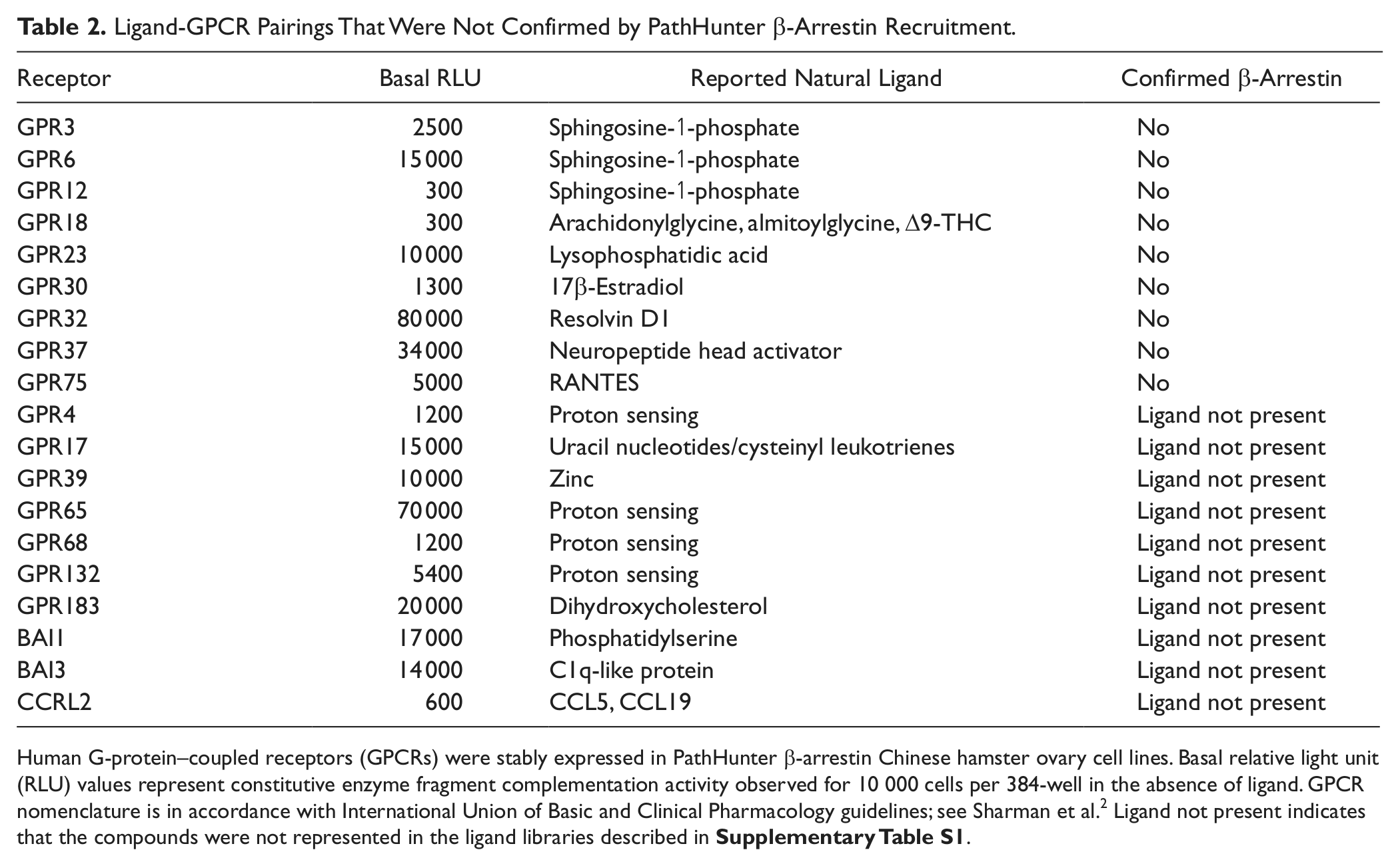
Human G-protein–coupled receptors (GPCRs) were stably expressed in PathHunter β-arrestin Chinese hamster ovary cell lines. Basal relative light unit (RLU) values represent constitutive enzyme fragment complementation activity observed for 10 000 cells per 384-well in the absence of ligand. GPCR nomenclature is in accordance with International Union of Basic and Clinical Pharmacology guidelines; see Sharman et al.
2
Ligand not present indicates that the compounds were not represented in the ligand libraries described in
Identification of Putative Surrogate Agonists at MRGPRX2, GPR88, and GPR97
The utility of β-arrestin recruitment as a screening technology was further demonstrated by the identification of a number of novel surrogate ligand-receptor pairings, including receptors that have previously proven difficult to partner. MRGPRX2 has been implicated in sensory and host defense mechanisms and can respond to a large number of ligands encompassing both peptides and small molecules.21,22 Here, β-arrestin recruitment confirmed the activity of the known peptide ligands cortistatin and somatostatin and further identified the delta-1 opioid receptor agonist TAN-6 as a novel agonist at MRGPRX2 ( Fig. 3 A , B ). 23 Perhaps coincidentally, MRGPRX1 was previously shown to share ligands with the opioid receptors. For example, naloxone can block responses via both MRGPRX1 and opioid receptors by binding to opioid receptors alone. 24 In the present study, the delta opioid receptor antagonists BNTX and SDM25N were unable to block MRGPRX2 arrestin recruitment induced by TAN-67 (data not shown), and hence it is likely that the action of TAN-67 is direct rather than a transactivation event mediated via opioid receptors.
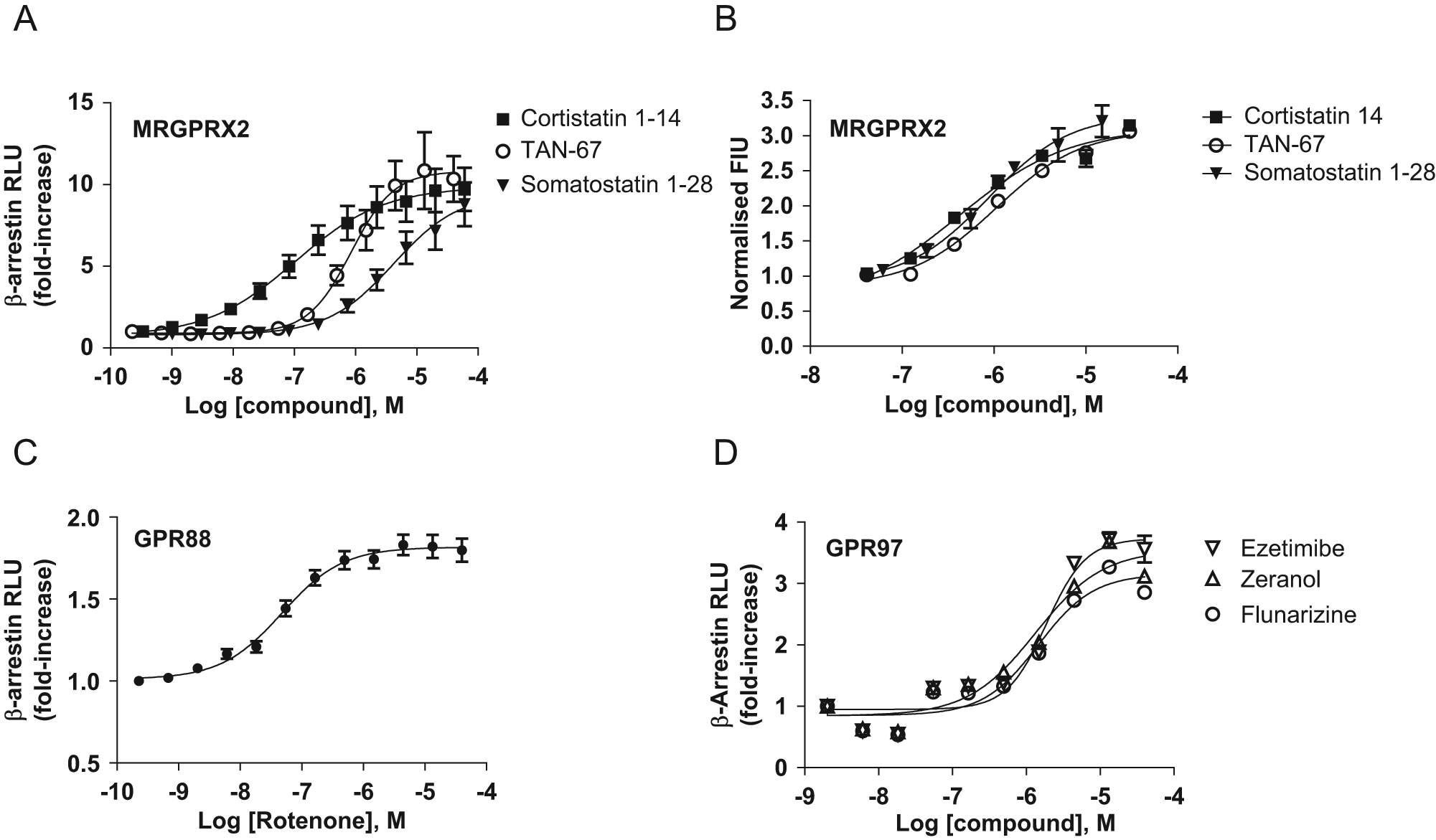
Novel surrogate ligand identification for the receptors MRGPRX2, GPR88, and GPR97. Dose-response curves for TAN-67 and the endogenous ligands cortistatin-14 and somatostatin 1-28 performed using PathHunter β-arrestin MRGPRX2 cells. (
GPR88 was identified as a novel GPCR specifically located within the striatum of the rat brain and was thus initially designated Strg or striatum-specific GPCR. The unique expression profile of GPR88 would suggest a potential role in the striatum and basal ganglia, and as such, dysfunction associated with GPR88 may play a role in neuropsychiatric and neurodegenerative disease.25,26 In the present study, we did not identify candidates that could be considered to act as endogenous ligand for GPR88. However, we did observe that the mitochondrial complex I inhibitor, rotenone, promoted β-arrestin recruitment to GPR88 with an EC50 of 100 nM ( Fig. 3C ). Rotenone is recognized for its ability to disrupt the mitochondrial electron transport chain, giving rise to oxidative stress with deleterious effects, and hence it is notable that rotenone positively promoted β-arrestin recruitment to GPR88 throughout the 90-min incubation period. 27 The potency and selectivity of rotenone for GPR88 were determined in dose-response studies performed using 12 unrelated PathHunter GPCRs, including SSTR2 (data not shown). Rotenone did not cause constitutive EFC activity to fall below basal levels in any orphan receptor cell line tested, which could have been considered a sign of overt toxicity. A series of synthetic agonists acting at GPR88 have recently been shown to promote a decrease in intracellular cyclic AMP via coupling through Gαi subunits. 28 At the present time, we have not been able to demonstrate G-protein–coupled signaling via calcium mobilization in response to rotenone following the co-transfection of Gqi5, Gα16, and Gα16z49e promiscuous G-protein subunits. Further work will be required to elucidate the mechanism of rotenone-induced GPR88 receptor signaling, but it is of interest to consider the impact of the modulation of GPR88 by rotenone on neuronal function and survival.
GPR97 is a member of a novel subset of receptors that show similarity to the class B GPCR family yet are structurally distinct in that they comprise a large, multidomain N-terminal extracellular region and hence are known as the LNB receptors. 29 The physiological roles of the LNB receptors are largely unknown but may combine the functions of extracellular adhesion molecules and GPCRs to mediate intercellular and cell-matrix interactions. As such, the LNB receptors are thought to play a role in immune function, angiogenesis, cell polarity, growth, and development. Limited data exist to indicate that LNB receptors may indeed couple through conventional G-protein signaling mechanisms, and hence it is noteworthy that here each of the LNB receptor PathHunter β-arrestin cell lines studied (GPR97 BAI1, BAI2, BAI3) gave rise to a basal EFC signal. β-Arrestin recruitment may thus be common to this receptor class and may be of value in elucidating LNB receptor biology. Three surrogate ligands were found to be active at GPR97 ( Fig. 3D ). The inhibitor of cholesterol absorption ezetimibe, the nonselective calcium channel blocker and histamine H1 blocker flunarizine, and the nonsteroidal estrogen zeranol, all in the 1- to 10-µM range, promoted β-arrestin recruitment to GPR97 but no other GPCRs tested. Our preliminary data based on co-transfection of Gqi5, Gα16, and Gα16z49e G-protein subunits indicated that this activity appeared to be G-protein independent. Limited structure-activity relationships around these surrogate ligands confirmed their agonist activity, but potency was not further increased (data not shown). Although the nature of the interaction between these novel surrogate ligands and GPR97 is not presently clear, these results would indicate that the LNB group of receptors may indeed be tractable in terms of delivering screening assays and small-molecule modulators for drug discovery. Recent studies demonstrating G-protein–mediated constitutive activity in a subset of adhesion GPCRs and specific coupling of GPR97 to Gqo3 would support this notion. 30
The rationale behind the present study was to investigate whether a potential bias toward β-arrestin might facilitate the identification of novel ligands for the orphan GPCRs that may not have been detected by more classical G-protein signaling assays employed in previous deorphanization campaigns.3–5 A variety of arrestin-based assay formats are now established to enable the monitoring of ligand-induced GPCR activation, many of which are amenable to HTS. The cytosolic redistribution of green fluorescent protein (GFP)–tagged arrestin may be monitored as an indirect readout of receptor-arrestin interaction (Transflour; Molecular Devices), but this requires high-content analysis and does not represent a specific readout for the receptor of interest. Nonimaging assay technologies requiring only conventional luminescence readers are based on dual tags incorporated into both receptor and arrestin molecules. Thus, upon receptor-arrestin interaction, β-galactosidase complementation (PathHunter; DiscoverX) or β-lactamase reporter activation (Tango; Invitrogen, Life Technologies) provides enzymatic readouts of ligand-induced receptor activation. β-Arrestin–receptor interaction may also be studied using bioluminescence (BRET)- and fluorescence resonance energy transfer (FRET) techniques, which are well suited to measure changes in the proximity between fluorescently tagged partner proteins in living cells and on a millisecond timescale.
The PathHunter assay format employed here is based on a 1:1 interaction between target receptor and β-arrestin in the reconstitution of reporter enzyme activity. In contrast, G-protein signaling-based assays may have the advantage of increased sensitivity resulting from amplification of the signaling cascade. Although advantageous for HTS, particularly in conjunction with the high receptor density attained in recombinant expression systems, this must be balanced by the potential to identify ligand-receptor interactions that do not translate into native tissue. 31 In the absence of signal amplification, the PathHunter assay format may thus give rise to fewer false positives. Each of the arrestin assay formats mentioned above offers opportunities to address GPCR function in academic and pharmaceutical settings. Our preliminary work demonstrated that PathHunter β-arrestin recruitment technology gave rise to reproducible, HTS-compatible screening assays. In the present study, the availability of stable cell lines for nearly all the remaining orphan GPCRs gave rise to a unique opportunity to undertake a comprehensive deorphanization campaign based on an alternative to G-protein–mediated readouts.
β-Arrestin screening was able to identify novel ligands and provide additional information for a number of receptors, but despite maintaining a robust screening process, our approach did not reveal novel endogenous ligands for the majority of orphan receptors studied ( Table 3 ). Furthermore, of the novel ligands we were able to identify, none displayed a bias or selectivity toward β-arrestin signaling. This serves as an intriguing reminder as to why these receptors have remained orphans, despite the continued application of emerging technologies by many research groups. This outcome could be explained by the possibility that the ligand banks we employed did not comprehensively include the required cognate ligands. The goal of our screening activity was focused on the detection of novel endogenous agonists, and it must be acknowledged that weak or partial agonists may not have been detected. Although there was an apparent paucity of ligand-receptor interactions, it should also be considered that compounds within our ligand libraries could alternatively display antagonist activity. Exploration of this possibility, however, would require reformatting of the assay screen and the availability of suitable agonists for the orphan receptors.
Ligand Screening for Orphan GPCRs Using PathHunter β-Arrestin Recruitment.
Orphan human G-protein–coupled receptors (GPCRs) stably expressed in PathHunter β-arrestin Chinese hamster ovary (CHO) or human embryonic kidney (*) cell lines. GPR19 was transiently expressed in a PathHunter β-arrestin CHO cell line. Basal relative light unit (RLU) values represent constitutive enzyme fragment complementation activity observed for 10 000 cells per 384-well. GPCR nomenclature is in accordance with International Union of Basic and Clinical Pharmacology guidelines; see Sharman et al. 2
In addition to GPCR desensitization and internalization, the arrestins have now emerged as distinct, G-protein–independent signaling molecules with the ability to interact with a wide variety of intracellular signaling and cytoskeletal elements. 6 As well as switching receptors off, arrestins provide additional mechanisms by which GPCRs may diversify their signaling output. Thus, ligands exhibiting a preferential bias toward arrestin signaling as well as arrestin-independent, G-protein–biased signaling are likely to be of value in the development of new therapeutics.
Our hypothesis that arrestin-biased signaling might hold the potential to unlock long sought-after ligand pairings was not proven to be the case within the constraints of the ligand sets and engineered receptor systems examined. Indeed, where this phenomenon does exist, it may be more applicable to surrogate rather than cognate ligands. Successful GPCR deorphanization must address a number of biological variables. The orphan receptor should be expressed, as far as possible, in its native environment with access to required accessory proteins and/or oligomerization partners, and it is currently not known if a CHO or HEK cell background would provide the array of accessory components required for full functionality. A ligand bank or tissue extract must contain the cognate ligand at an effective concentration, and an appropriate assay readout must be configured to detect the signaling pathway elicited by the specific ligand-receptor interaction. The PathHunter reagents used throughout this study were of high functional quality, as evidenced by the reconstitution of a detectable basal or constitutive level of β-arrestin recruitment. It is presumed that such constitutive recruitment is driven by the inherent constitutive activity of each GPCR, a phenomenon that has gained increasing recognition. 32 The extent to which this observation may be nonreceptor driven and derived through the recombinant expression systems employed should be considered. However, we observed constitutive β-arrestin recruitment for each of 82 GPRs studied, which would also indicate that this phenomenon is a feature common to most GPCRs.
Although our ligand sets were generated to provide an extensive range of ligand types, it is impossible to be completely comprehensive in this respect. The reasons as to why there remain a large number of orphan GPCRs are likely to be complex, and whether large-scale screening programs will continue to represent an appropriate way forward is an interesting point for debate. The future of deorphanization may instead progress on a smaller scale, based on a more focused effort informed by tissue distribution, genetic ablation, and disease association. Although arrestin recruitment is a broadly applicable GPCR technology, additional detection formats may be required for receptors and ligand-receptor pairings that may be both G-protein and β-arrestin independent. 33 Our data, however, strongly support the concept that PathHunter β-arrestin recruitment represents a robust, convenient, and receptor-specific screening platform to address GPCR biology and is amenable to both high-throughput deorphanization programs and the interrogation of GPCR pharmacology.
Finally, although we were unable to definitively identify numerous novel, endogenous ligands, recent reports from two independent groups continue to illustrate that novel ligand-receptor pairings are still to be discovered. Guided by the known role for the orphan receptor GPR183, also known as Epstein-Barr virus–induced gene 2, in the adaptive immune response, Hannedouche et al 34 and Liu et al 35 identified 7α,25-dihydroxycholesterol as a novel GPR183 agonist. A considerable research effort was required to purify this novel, potent, and selective ligand from primary tissue sources. Unfortunately, however, 7α,25-dihydroxycholesterol was not present within our ligand library sets. It has been proposed that orphan GPCRs may exist, which signal independently of an endogenous ligand or may function as constituent of a GPCR heterodimer. However, with more focused research efforts and the continued application of emerging GPCR screening technologies, it is probable that novel endogenous ligands are yet to be discovered.
Footnotes
Acknowledgements
We acknowledge Alan Wise for the original idea and Mike Snowden, Pyare Khanna, Sanj Kumar, and Justin Bryans for initiating and supporting the collaboration. We thank colleagues at GSK: Amanda Jowett and Chris Plumpton for monoclonal antibody generation, Sian Lewis for calcium signaling experiments, and Darren Green for chemistry advice; colleagues at MRCT: Ahmad Kamal, Emilie Bureau, and Preeti Bakrania, for secondary assay support; and colleagues at DiscoveRx: Phil Achacoso, Chin-Yee Loh, and Albert Doan for help in generating and characterizing the cell lines.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The project was funded by the MRC Development Gap Fund, grant number A853-0100.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.