
Abstract
The secretory and transmembrane isoforms of prostatic acid phosphatase (PAP) can dephosphorylate extracellular adenosine 5′-monophosphate (AMP) to adenosine, classifying PAP as an ectonucleotidase. Currently, there are no compounds that inhibit PAP in living cells. To identify small-molecule modulators of PAP, we used a 1536-well–based quantitative high-throughput fluorogenic assay to screen the Library of Pharmacologically Active Compounds (LOPAC1280) arrayed as eight-concentration dilution series. This fluorogenic assay used difluoro-4-methylumbelliferyl phosphate as substrate and collected data in kinetic mode. Candidate hits were subsequently tested in an orthogonal absorbance-based biochemical assay that used AMP as substrate. From these initial screens, three inhibitors of secretory human (h) and mouse (m)PAP were identified: 8-(4-chlorophenylthio) cAMP (pCPT-cAMP), calmidazolium chloride, and nalidixic acid. These compounds did not inhibit recombinant alkaline phosphatase. Of these compounds, only pCPT-cAMP and a related cyclic nucleotide analog (8-[4-chlorophenylthio] cGMP; pCPT-cGMP) inhibited the ectonucleotidase activity of transmembrane PAP in a cell-based assay. These cyclic nucleotides are structurally similar to AMP but cannot be hydrolyzed by PAP. In summary, we identified two cyclic nucleotide analogs that inhibit secretory and transmembrane PAP in vitro and in live cells.
Introduction
Prostatic acid phosphatase (PAP, also known as ACPP) has been used for more than 50 y as a prostate cancer biomarker. 1 Recently, we found that the transmembrane isoform of PAP was expressed in nociceptive (pain-sensing) dorsal root ganglia neurons. 2 In addition, we found that the secretory and transmembrane isoforms of PAP have ectonucleotidase activity, as demonstrated by the ability of each isoform to dephosphorylate extracellular adenosine 5′-monophosphate (AMP) to adenosine. In mice, intrathecal injection of the secretory isoform of PAP has antinociceptive effects in chronic inflammatory and neuropathic pain models that persist for 3 d. 2 These antinociceptive effects were entirely dependent on adenosine A1 receptor activation, suggesting PAP generates adenosine in vivo.
Intrathecal injections are routinely performed in humans, but they are invasive. Moreover, it is impractical to deliver PAP protein orally because, like all proteins, PAP is susceptible to proteolysis in the digestive tract. In contrast, small-molecule activators of PAP could enhance the activity of endogenously expressed PAP and be made orally bioavailable. At present, there are no known small-molecule activators of PAP.
Conversely, it would be advantageous to have small molecules that selectively and acutely inhibit PAP for physiological studies. L-(+)-tartrate is the most commonly used inhibitor of PAP but must be used at high (mM) concentrations. Also, L-(+)-tartrate inhibits other acid phosphatases. 3 α-Benzylaminobenzylphosphonic acid (BABPA), a derivative of benzylphosphonic acid, was found to inhibit PAP in the low-nanomolar range. 4 Although potent, this inhibitor is not routinely used because it is difficult to obtain (not commercially available and must be synthesized). It is unknown if BABPA inhibits other phosphatases. Discovering selective PAP inhibitors would provide a way to examine the acute biological effects of PAP inhibition and could complement studies with Pap-/- mice where enzyme activity is eliminated throughout life. 2
We previously identified several structurally diverse inhibitors of human PAP (hPAP) using a high-throughput 384-well single-point fluorogenic screen. 5 Here, we modified this single-point assay so that we could perform a quantitative (multiconcentration) high-throughput screen in 1536-well plates with secretory hPAP and difluoro-4-methylumbelliferyl phosphate (DiFMUP) as substrate. Quantitative high-throughput screening (HTS) has several advantages over traditional single-point screens, including fewer false-negatives and generation of dose-response curves as part of the screen. 6 We screened the Library of Pharmacologically Active Compounds (LOPAC1280) as an eight-point dilution series and identified several potential hPAP inhibitors. Three of these compounds also inhibited hPAP and recombinant mouse PAP (mPAP) in an orthogonal biochemical assay. This orthogonal assay used AMP, a physiologically relevant PAP substrate. Of these compounds, pCPT-cAMP and a related analog (pCPT-cGMP) were the only compounds that inhibited PAP in a real-time cell-based calcium mobilization assay. These cyclic nucleotide analogs are the first known compounds that inhibit PAP in vitro as well as in living cells.
Materials and Methods
Reagents
Most reagents were purchased from Sigma-Aldrich (St. Louis, MO), including HEPES, DMSO, sodium citrate, malachite green oxalate, sodium molybdate, sodium L-(+)-tartrate, sodium orthovanadate, sodium fluoride, EDTA, HCl, AMP, Triton X-100, Tween-20, purified hPAP (#P1774), recombinant bovine alkaline phosphatase (ALP; #P8361), and human protein tyrosine phosphatase-1B (PTP; #P6244). DiFMUP was obtained from Invitrogen (Carlsbad, CA), and potato acid phosphatase (pAP) was obtained from Roche Applied Science (Indianapolis, IN). Recombinant mPAP was generated as described previously. 7 Medium binding black solid-bottom 1536-well plates were obtained from Greiner Bio One (Monroe, NC) and were used for the LOPAC screen. Black clear-bottom 96-well plates that were used to measure hydrolysis of AMP were purchased from Corning Incorporated (Corning, NY). The buffer used for hPAP and mPAP fluorogenic assays was 50 mM HEPES, pH 7.0, 1 mM EDTA, and 0.01% Tween-20. A buffer consisting of 50 mM sodium acetate, pH 5.3, 0.01% Tween-20 was used for the pAP fluorogenic assay, whereas ALP was assayed in 50 mM Tris-HCl, pH 8.0, 0.01% Tween-20.
The LOPAC1280 library and dry powder versions of the selected hit compounds identified from the LOPAC1280 screen were obtained from Sigma-Aldrich. The LOPAC1280 library compounds were arrayed as an interplate dilution series starting from 10 mM stock in DMSO as described elsewhere. 8 6-Hydroxy-5-nitro-2-[(E)-2-(2-propoxy-naphthalen-1-yl)-vinyl]-3H-pyrimidin-4-one (Asinex 49) was purchased from Asinex Corporation (BAS 08865249; Moscow, Russia).
BABPA was synthesized based on a published procedure. 4 Briefly, to a stirring solution of E-N-benzylidene-1-phenylmethanamine (0.5 g, 2.56 mmol) at 0 °C was added triethyl phosphite (0.448 g, 2.56 mmol). The reaction mixture was heated to 70 °C for 12 h, at which time any excess triethyl phosphite was removed under reduced pressure. The remaining residue was purified directly on silica. Gradient elution (40–70% ethyl acetate in hexanes) afforded the desired product as a colorless, viscous oil: yield 554 mg, 1.66 mmol, 65%.
To a stirring solution of diethyl (benzylaminophenyl) methyl phosphonate (0.13 g, 0.39 mmol) in water was added hydrochloric acid (1 mL, 10 mmol). The reaction mixture was heated to 50 °C for 4 h. Upon completion, the reaction mixture was carefully neutralized with sat. aq. sodium bicarbonate. The solution was filtered and purified directly by reverse-phase chromatography. Gradient elution (10–60% acetonitrile in water) and subsequent lyophilization of the appropriate fractions afforded the desired product as a colorless, powdery solid: yield 0.027 g, 0.098 mmol, 25%.
Quantitative HTS Assay Protocol and HTS Data Analysis
To conduct the primary screen against the LOPAC library, 3 µL of enzyme (final concentration: 2 nM for hPAP) in columns 1, 2, and 5 to 48 and 3 µL of the assay buffer in columns 3 and 4 were dispensed into 1536-well Greiner black assay plates. Compounds (23 nL) were transferred via Kalypsys pintool equipped with 1536-pin array (10 nL slotted pins; V&P Scientific, San Diego, CA), with the LOPAC compounds pin-transferred into columns 5 to 48 and the control compound, BABPA, pin transferred into column 2. The plates were incubated for 15 min at room temperature before the addition of 1 µL fluorogenic substrate DiFMUP (final concentration 100 µM). Throughout the screen, all reagent bottles were kept at 4 °C to minimize degradation. Immediately following substrate addition, fluorescence data were collected on a ViewLux high-throughput imager (PerkinElmer, Waltham, MA) every minute for 3 min using a standard UV excitation filter (340 nm, bandwidth 60 nm) and the umbelliferone emission filter of 450 nm (bandwidth 20 nm); the change in fluorescence, measured for every sample over the 3-min initial reaction time course, was used to calculate the Z′ statistical parameter using the formula in Zhang et al., 9 as well as for calculation of normalized responses. Data were normalized against no-enzyme wells (columns 3 and 4) and enzyme-containing wells (column 1) and were further fitted using a four-parameter Hill equation through publicly-available curve-fitting algorithms (http://ncgc.nih.gov/pub/openhts/), as described in detail elsewhere.6,8
For follow-up fluorogenic assays against mPAP and additional phosphatases, the protocols were the same as described above with the exception that the compounds were arrayed as 12-point dose responses within each plate. Final concentrations of the phosphatases used in the profiling were as follows: 1.8 nM for mPAP, 1.5 nM for pAP, and 5 pM for ALP. The data were normalized against enzyme-containing and no-enzyme controls, and data were fitted using a sigmoidal dose-response regression algorithm in GraphPad Prism (La Jolla, CA).
AMP Hydrolysis Enzyme Assay
Enzyme assays were performed as previously described. 7 Briefly, enzyme (1 U hPAP, 1 U mPAP or 100 U ALP) was added to reaction mixture (50 µL total volume) in a 1.5 mL microcentrifuge tube containing 400 µM, 1 mM, or 100 µM AMP corresponding to the KM of hPAP, mPAP, and ALP for AMP, respectively, 50 mM HEPES buffer pH 7.0 and test compound (10–4 to 10–7 M). Compounds were preincubated with enzyme for 3 min at 37 °C prior to the addition of 950 µL malachite green color reagent (0.03% [w/v] malachite green oxalate, 0.2% [w/v] sodium molybdate, 0.05% [v/v] Triton X-100 in 0.7 M HCl). Reactions were incubated at room temperature for 30 min, and the colorimetric reaction was quenched with 22.4 µL of 38% sodium citrate. The samples (100 µL) were transferred to a 96-well black, clear-bottom plate (Corning), and inorganic phosphate release was quantified at 650 nm on a SpectraMax Plus plate reader (Molecular Devices, Sunnyvale, CA). Negative controls contained AMP, buffer, and no enzyme. For each compound tested, negative controls also contained the same concentration of compound as being tested. The absorbance measurements in the no-enzyme control were subtracted from the absorbance measurements for each test sample. The raw absorbance data were converted to nmol of phosphate released per minute using an inorganic phosphate standard curve (KH2PO4). Data were analyzed using EXCEL, and the dose-response data were fit by a nonlinear regression equation using GraphPad Prism 5.
Calcium Mobilization Assay
Calcium imaging was performed as described previously. 10 Briefly, HEK293 cells were plated at 2 × 106 per glass-bottom MatTek dish (MatTek, Ashland, MA). Prior to plating, each dish was coated with 0.1 µg/mL poly-D-lysine (Sigma). After 24 h, cells were transfected using lipofectamine (Invitrogen) per manufacturer’s instructions. Control experiments contained pcDNA3.1/chimeric Gαq-s5 protein/pcDNA3.1/Venus (0.3/0.3/0.3/0.1 µg DNA ratio). Experimental conditions used A2B/chimeric Gαq-s5 protein/pcDNA3.1 or transmembrane PAP/Venus. The following day, cells were washed three times with HBSS (Gibco) loaded with 2 µM Fura-2AM (Invitrogen) in HBSS for 1 h at room temperature in the dark and were washed three times with HBSS and incubated for 30 min prior to imaging. Cells were preincubated with each compound in HBSS for 1 h and for 5 min with 10 µM αβ-methylene adenosine diphosphate (ADP) in HBSS to inhibit endogenous ecto-5′-nucleotidase. A 30 s baseline was obtained followed by 2 min of agonist in the presence of 10 µM αβ-methylene ADP. Cells were imaged on a Nikon Ti-E (Nikon, Melville, NY) and analyzed using NIS Elements Imaging software. Data were exported to EXCEL to create graphs and GraphPad Prism 5 to analyze area under curve. Area under curve was obtained for 1 min postagonist.
Results and Discussion
BABPA Inhibits Mouse and Human Secretory PAP
BABPA inhibits PAP with nanomolar efficacy. 4 As a positive control, we confirmed that BABPA inhibited hPAP (IC50 = 4.97 nM) in our fluorgenic assay ( Fig. 1A ). In addition, BABPA inhibited mPAP (IC50 = 20.6 nM) but not pAP or ALP ( Fig. 1A ). Taken together, these data indicate BABPA is relatively selective for mammalian PAP.
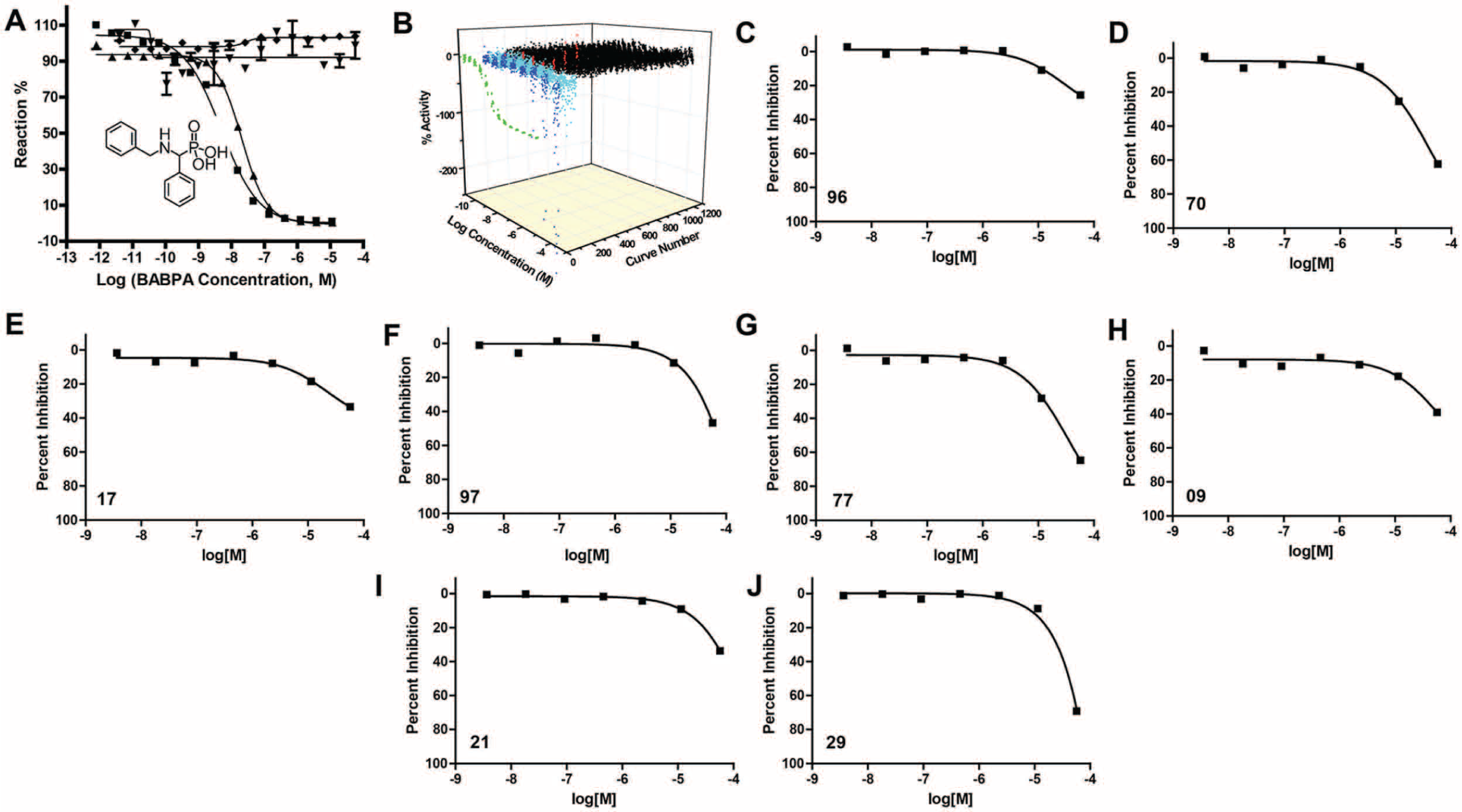
Primary screen to identify prostatic acid phosphatase (PAP) inhibitors. (
Quantitative HTS of the LOPAC1280 Library
We next used quantitative HTS to identify candidate PAP inhibitors in the LOPAC1280 library. 6 This entailed testing the library compounds in a dose-response format, with eight concentrations ranging from 0.3 nM to 115 µM. The mean Z′ value 9 was ~0.9 across all plates, indicating a stable assay performance. After screening the library, the compounds were categorized into four groups: inactive, false-positives, potential inhibitors, and potential activators ( Fig. 1B ). The groups were based on the shape and quality of the dose-response curves obtained (IC50, presence of two asymptotes, partial versus complete curve). Autofluorescent hits were excluded when the starting fluorescence intensity in the kinetic fluorogenic assay was greater than that of the uninhibited enzyme control. Based on these results, a set of 13 compounds, composed of 8 potential inhibitors ( Fig. 1C – J ; Table 1 ) and 5 potential activators, was selected for confirmatory testing and selectivity profiling against mammalian and nonmammalian phosphatases.
IC50 and Max Response for Eight Candidate Inhibitors against hPAP, mPAP, pAP, and ALP in the Fluorogenic and 5′-AMP Hydrolysis Assays (All In Vitro) a .
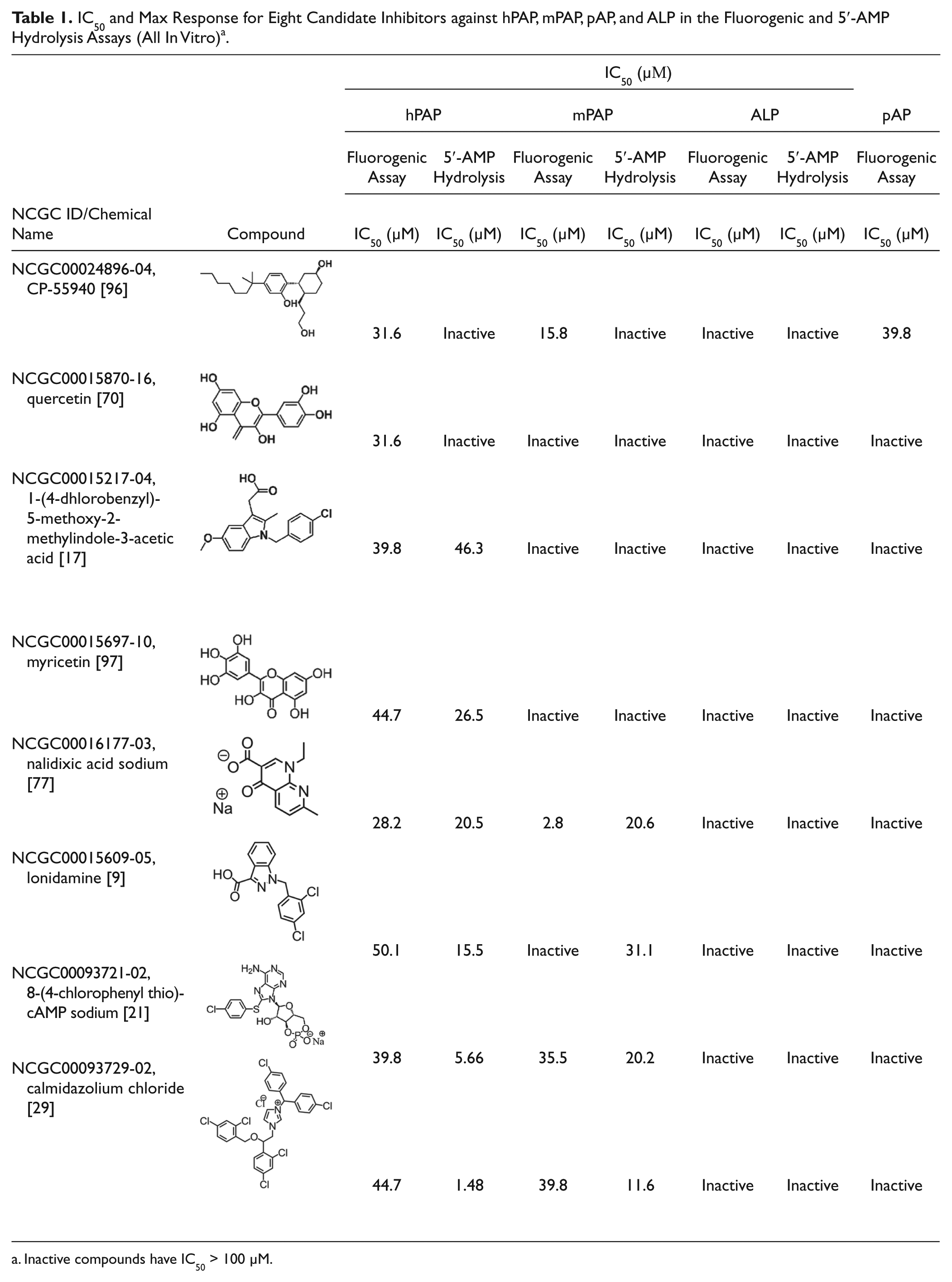
Inactive compounds have IC50 > 100 µM.
Of the eight potential inhibitors, pCPT-cAMP, calmidazolium chloride, and nalidixic acid also inhibited mPAP but not pAP or ALP using the fluorogenic substrate ( Table 1 ). Although all five activators enhanced the activity of hPAP in our initial screen and in confirmatory fluorogenic assays, none activated hPAP in our orthogonal AMP hydrolysis assay (described below) and hence were not studied further.
Confirmation of PAP Inhibitors Using an Orthogonal 5′-AMP Hydrolysis Assay
To independently validate hits and determine if any were false-positives (i.e., nonspecific compound interactions with the DiFMUP substrate or quenching), we employed an orthogonal non–fluorescence-based enzyme assay that used AMP, an endogenous PAP substrate. 7 For each reaction, hydrolysis of AMP by hPAP was measured in the presence of the eight inhibitors described above (at increasing doses), while the substrate concentration was held constant at a value close to the reported KM for each enzyme (hPAP: 400 µM AMP [KM = 0.3–2 mM], mPAP: 1 mM AMP [KM = 0.9–1.6 mM], ALP: 100 µM AMP [KM = 0.018–15.4 mM]).11,12 Of these eight compounds, calmidazolium chloride, pCPT-cAMP, nalidixic acid, and lonidamine inhibited hPAP and mPAP in the low-micromolar range ( Table 1 ; Fig 2A – D ; calculated IC50 values and structures are inset).
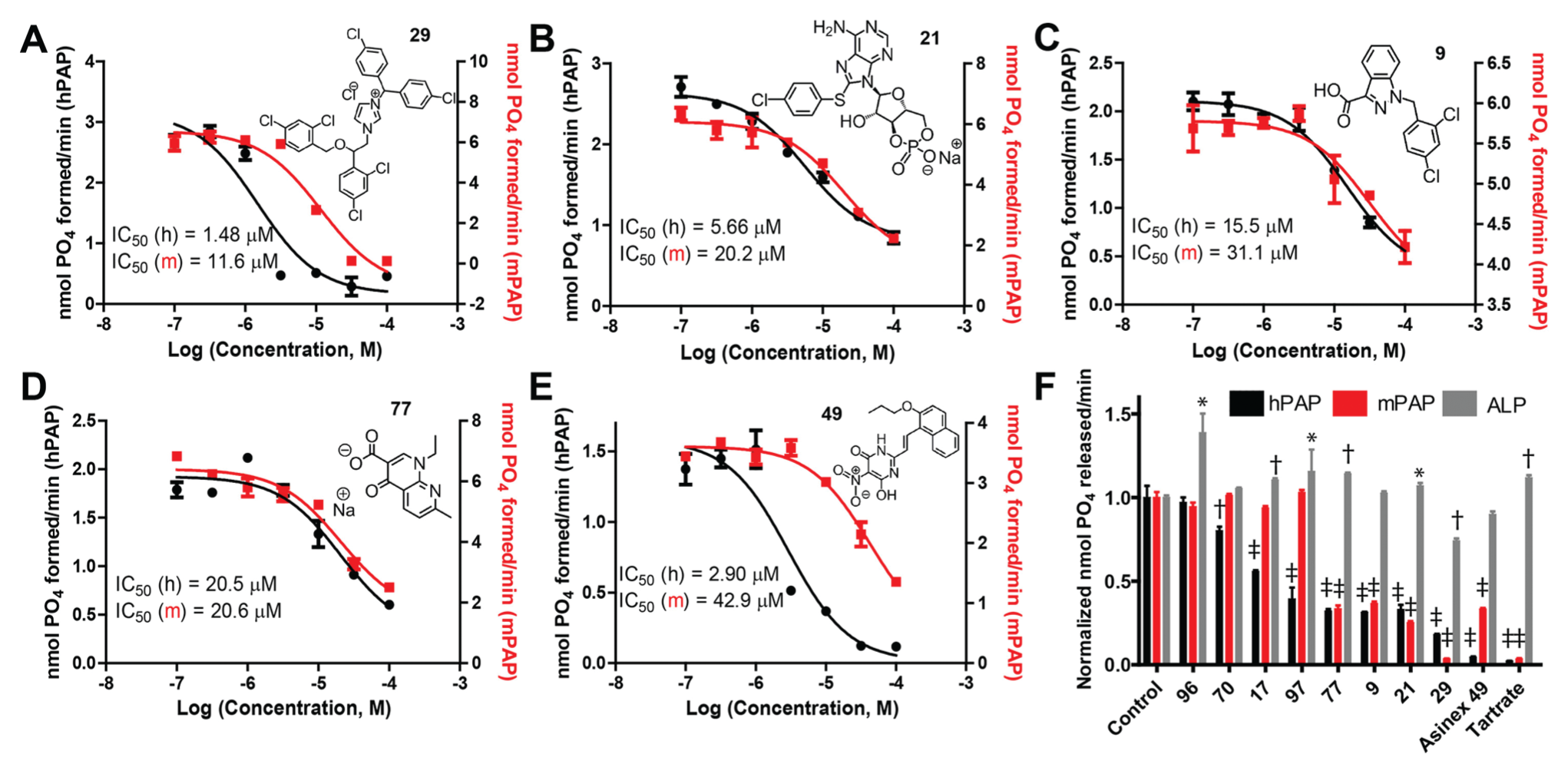
Dose-response curves for screen-identified inhibitors in an orthogonal adenosine 5′-monophosphate (5′-AMP) hydrolysis assay. (
In addition, we tested compound 49 [6-hydroxy-5-nitro-2-[(E)-2-(2-propoxy-naphthalen-1-yl)-vinyl] -3H-pyrimidin-4-one], a low-micromolar hPAP inhibitor previously identified in a fluorogenic-based screen. 5 Compound 49 inhibited AMP hydrolysis by hPAP and mPAP with an IC50 of 2.90 µM for hPAP and 42.9 µM for mPAP ( Fig. 2E ) but did not inhibit ALP ( Fig. 2F ). As a second inhibitor control, we used L-(+)-tartrate (10 mM), which has a low affinity for PAP and must be used at high concentrations to inhibit PAP enzymatic activity. 3 L-(+)-tartrate inhibited hPAP and mPAP in our 5′-AMP hydrolysis assay but did not inhibit ALP ( Fig. 2F ).
Transmembrane PAP Ectonucleotidase Activity Was Inhibited in a Real-Time Cell-Based Assay
We recently developed a real-time, cell-based calcium mobilization assay to study the hydrolysis of AMP to adenosine by transmembrane PAP. 10 This assay is based on our observation that the A2B adenosine receptor can be activated by adenosine but not AMP. HEK293 cells were transiently transfected with the A2B adenosine receptor and the chimeric G protein subunit (Gq-s5), which couples the A2B adenosine receptor to calcium mobilization (visualized with Fura-2AM). Transmembrane PAP was also transfected in some conditions ( Fig. 3 ). Hydrolysis of AMP to adenosine was measured with and without inhibitors, as read out by an increase in calcium mobilization. A dose response was generated for BABPA ( Fig. 3A ) and pCPT-cAMP ( Fig. 3B ). BABPA (5 µM) and pCPT-cAMP (100 µM) did not block activation of the A2B receptor (using adenosine as ligand) but dose dependently inhibited hydrolysis of AMP to adenosine by transmembrane PAP ( Fig. 3A , B ). In contrast, nalidixic acid blocked A2B receptor activation by adenosine (data not shown), which precluded us from studying nalidixic acid in this cell-based assay. In addition, we were unable to study calmidazolium chloride and lonidamine in this cell-based assay because both compounds killed the cells.
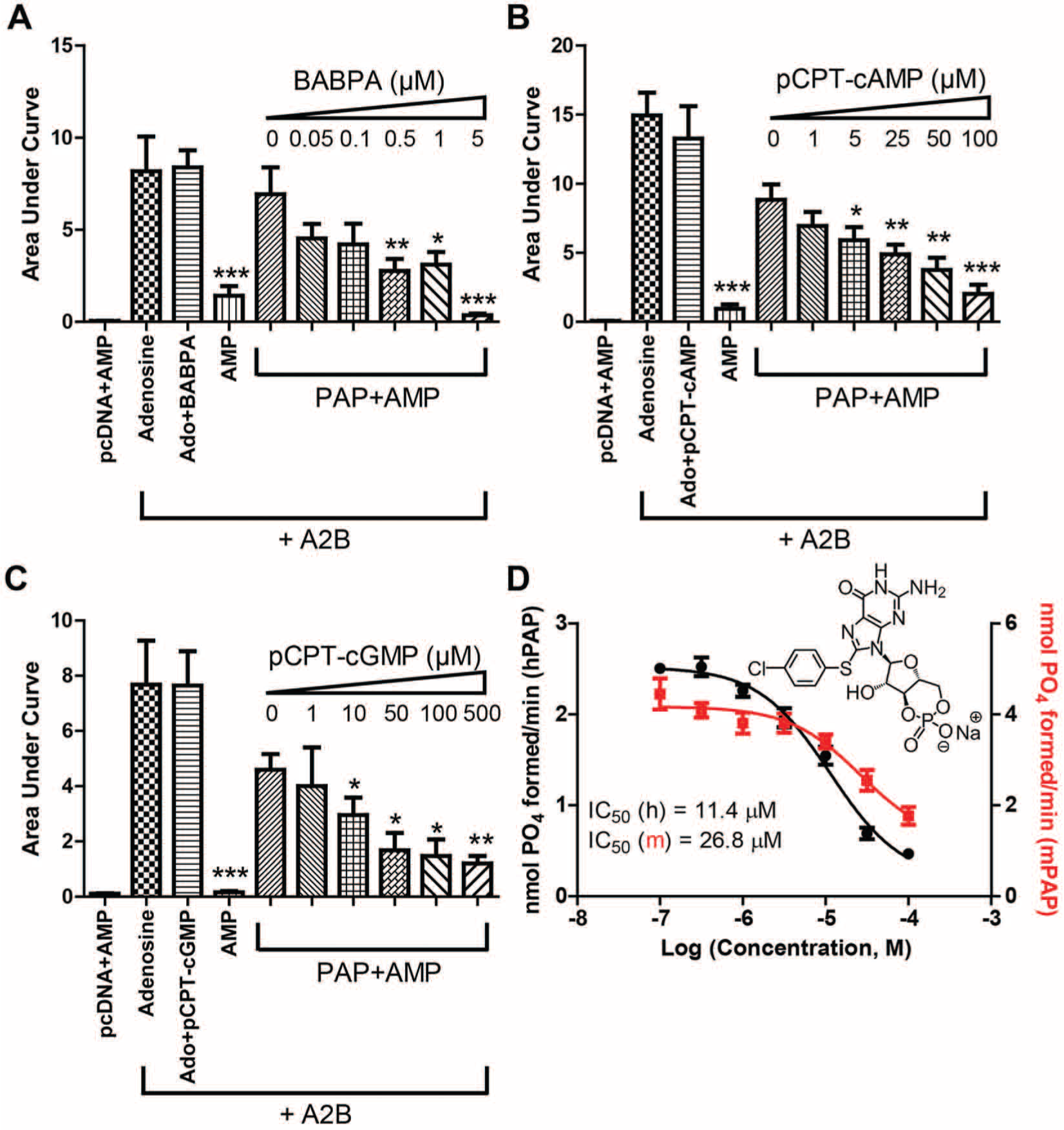
Dose responses for BABPA, pCPT-cAMP, and pCPT-cGMP in a cell-based calcium mobilization assay. (
Given that pCPT-cAMP was the only compound that inhibited human and mouse PAP in vitro and in cells, we next tested a related cyclic nucleotide analog, 8-(4-chlorophenylthio) cGMP (pCPT-cGMP). We found that pCPT-cGMP also inhibited transmembrane PAP in the cell-based calcium mobilization assay ( Fig. 3C ). In addition, pCPT-cGMP inhibited human and mouse secretory PAP in vitro using AMP as substrate ( Fig. 3D ) and using the fluorogenic substrate (IC50 = 31.1 µM, IC50 = 49.7 µM, respectively). Neither pCPT-cAMP nor pCPT-cGMP inhibited pAP, ALP, or PTP using the fluorogenic substrate (data not shown).
In this study, we used a fluorogenic assay that was capable of identifying small-molecule activators and inhibitors of PAP. PAP functions as an ectonucleotidase in nociceptive neurons. 2 Small-molecule activators and inhibitors could be used to study the acute effects of modulating PAP activity on nucleotide levels and adenosine production. After screening 1280 small molecules contained within the LOPAC1280 collection, we identified eight candidate inhibitors and five candidate activators. None of the activators identified from the primary screen enhanced PAP in an orthogonal AMP hydrolysis assay, suggesting these activators were false-positives. We were similarly unable to identify hPAP activators in an endpoint screen of 28 800 small molecules. 5 These results suggest that the odds of identifying hPAP activators are extremely low. In contrast, three of the candidate inhibitors (pCPT-cAMP, calmidazolium chloride, and nalidixic acid) identified in our present screen selectively inhibited hPAP and mPAP (but not pAP or ALP) in an orthogonal AMP hydrolysis assay. Of these three inhibitors, pCPT-cAMP was the only compound that inhibited PAP in live cells.
pCPT-cAMP is commonly used as a membrane-permeant activator of cAMP- and cGMP-dependent protein kinases and EPAC proteins, a class of cytoplasmic cAMP-dependent guanine-nucleotide-exchange factors. pCPT-cAMP is structurally similar to AMP, a physiologically relevant substrate of PAP. Although pCPT-cAMP is reportedly used to modulate intracellular signaling, our findings suggest pCPT-cAMP acts extracellularly to inhibit adenosine production in cells and tissues that express PAP. In fact, pCPT-cAMP is charged at physiological pH, raising the issue of how this compound enters cells. Our cell-based experiment indicates that pCPT-cAMP inhibits an extracellularly active enzyme (which is coupled to extracellular activation of a cell surface receptor), strongly arguing for an extracellular site of action. In addition, Waidmann and colleagues 13 found that pCPT-cAMP blocked equilibrative nucleoside transporter 1. Inhibition of this adenosine transporter elevated extracellular adenosine and activated adenosine A2A receptors in PC12 cells. 13
Although pCPT-cAMP effectively inhibited PAP, cAMP did not inhibit hPAP in the fluorogenic assay (data not shown). One explanation for why pCPT-cAMP but not cAMP inhibited PAP could be associated with the structural modification at the C8 position of the purine ring (chlorophenylthio group). Similarly, the related cyclic nucleotide analog pCPT-cGMP, which also contains the chlorophenylthio group at the C8 position, inhibited PAP in vitro and extracellularly in cells. Thus, the C8 modification may be necessary to inhibit PAP function. Intriguingly, these cyclic nucleotide analogs are structurally similar to AMP, the natural substrate of PAP, 2 but cannot be hydrolyzed to inorganic phosphate by PAP ( Figs. 2B , 3D ).
Nalidixic acid, calmidazolium chloride, and lonidamine inhibited PAP in biochemical assays; however, we were unable to assess the inhibitory activity of these compounds in cells because of off-target effects (nalidixic acid inhibited A2B whereas lonidamine and calmidazolium chloride killed cells). These compounds are thus not likely to be useful for inhibiting PAP in a more physiologically relevant setting, such as in live cells or in animals.
Lastly, high-throughput screens were used to identify inhibitors for tissue nonspecific alkaline phosphatase. 14 ALPs and PAP have ectonucleotidase activity and use AMP as a substrate. 15 Given that pCPT-cAMP inhibited PAP but not ALP, pCPT-cAMP may prove useful for selectively inhibiting PAP in future physiological studies.
Footnotes
Acknowledgements
We thank Joe Rittiner for help with the cell-based AMP hydrolysis assay.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by a grant to A.S. from the Molecular Libraries Initiative of the NIH Roadmap for Medical Research (U54MH084681), the Intramural Research Program of NHGRI, NIH, and by a grant to M.J.Z. from NINDS (R01NS067688).