
Abstract
The endocrine action of human (h) intestine-derived fibroblast growth factor 19 (hFGF19) toward liver cells necessitates a highly specific recognition system. We previously reported that at physiological concentrations (~30 pM), hFGF19 requires sulfated glycosaminoglycans (sGAGs) for its signaling via human FGF receptor 4 (hFGFR4) in the presence of a co-receptor, human βKlotho (hKLB), thus establishing specific targeting. Here we report that the specificity of hFGF19 signaling is greatly altered in a mouse model system. In in vitro cellular systems, at concentrations achievable in transgenic animals and in pharmacologic animal experiments (1–100 nM), hFGF19 activates mouse (m)FGFR1c, mFGFR2c, and mFGFR3c but not mFGFR4 in the presence of mKLB and nonheparin authentic sGAGs. Furthermore, in the presence of hepatic sGAGs or heparin, nanomolar hFGF19 activates mFGFR4, even in the absence of co-expressed mKLB. Taken together, these results indicate that the sGAG-assisted receptor specificity of hFGF19 signaling achieved in experimental mouse systems differs greatly from that in physiological human systems. This suggests the function and mechanism of hFGF19 signaling identified using mouse systems should be reevaluated.
Introduction
In human (h) and mouse (m), the fibroblast growth factor (FGF) family of ligands is encoded by 22 genes and can be divided into the endocrine FGF subfamily (hFGF19/mFGF15, FGF21 and FGF23), paracrine FGF subfamilies (FGF1 subfamily, including FGF1, FGF2, and FGF5; FGF3 subfamily, including FGF3, FGF4, and FGF6; FGF7 subfamily, including FGF7, FGF10, and FGF22; FGF8 subfamily, including FGF8, FGF17, and FGF18; FGF9 subfamily, including FGF9, FGF16, and FGF20), and intracellular FGF subfamily (FGF11, FGF12, FGF13, and FGF14).1–3 As a result of structural differences, the endocrine FGF subfamily ligands exhibit less affinity for heparin than the members of other subfamilies, and this is thought to be the molecular basis for their ability to function in an endocrine manner.
FGF19 is expressed in the distal small intestine in response to bile acid, and after secretion into the circulation, it reaches its target organ, the liver, via the portal vein. The circulating concentration of hFGF19 in healthy volunteers is reportedly in the picomolar range, with an average of approximately 30 pM. 4 In the liver, hFGF19 suppresses transcription of a key enzyme involved in bile acid synthesis to close a negative feedback loop. The mouse ortholog of the human FGF19 gene is designated Fgf15, reflecting its discovery history.1,5,6 Like hFGF19, mFGF15 is expressed in the distal small intestine and regulates bile acid synthesis in the liver. 7 hFGF19 also reportedly contributes to the regulation of blood glucose levels, along with other metabolic regulators, including FGF21 and insulin.3,8,9
The tyrosine kinase FGF receptor (FGFR) family is encoded by four genes that are translated into seven major groups of proteins containing a single membrane-spanning domain. With the exception of the intracellular FGF ligands, which do not interact with tyrosine kinase FGFRs, FGF ligands activate cell surface FGFRs with differential specificity. For paracrine FGF ligands to form active signaling complexes with the FGFRs, they must also bind sulfated glycosaminoglycans (sGAGs), which are sugar moieties consisting of variably sulfated, repeating disaccharide units that contribute to the composition of proteoglycans on the cell surface and to extracellular matrices. On the other hand, endocrine FGFs require the presence of a co-receptor, βKlotho (KLB) or αKlotho (KLA), 10 in addition to FGFR for their signal transduction. In humans, FGF23 specifically requires KLA, whereas hFGF19 and FGF21 require KLB.10–12 Although there are reports that KLA can also assist FGF19 signaling,13,14 we found that not to be the case when hFGF19 is at physiological concentrations (M. Nakamura and T. Imamura, unpublished observation). We previously reported that at physiological concentrations (pM levels), hFGF19 selectively activates hFGFR4 and requires the presence of hKLB and sGAGs. 15 Analogously, results from Fgf15 knockout mice indicate that mFGFR4 is the sole physiological receptor for mFGF15. 7 This finding, together with the early one that hFGF19 specifically binds to hFGFR4, 16 implies that hFGFR4 is the sole receptor for hFGF19. Furthermore, as mentioned, activation of hFGFR4 by hFGF19 at concentrations ranging from 3 pM to 500 pM was strongly dependent on the presence of sGAGs and on the co-expression of hKLB; thus, hFGF19 acts via a highly specific recognition system composed of four molecular entities: hFGF19, hFGFR4, hKLB, and sGAGs. The response-enhancing effects of the sGAGs were detected at concentrations of 0.3 µg/mL and higher, which is well within the physiological range. 15 However, several earlier studies using cells and animals of various species, although mainly mouse, suggested other FGFR subtypes (e.g., FGFR1c) can also mediate hFGF19 activity. Consequently, there remain several unanswered questions regarding the receptor specificity of hFGF19.
Normally, hFGF19 circulates at approximately 30 pM in the peripheral blood of healthy humans, but its mRNA expression is increased 20 to 200 times under some pathological conditions. 17 Interestingly, hFGF19 is present at 0.9 to 3.9 nM in the peripheral blood of Fgf19 transgenic mice 18 and perhaps two orders of magnitude higher when administered intraperitoneally at 2 mg/kg per day,17,19–21 which translates to 100 nM if we assume the animal’s body is composed entirely of water. This is noteworthy because hFGF19 exhibits potential utility for the treatment of metabolic diseases in humans,3,8,9 but efforts to apply hFGF19 clinically have been discouraged by the development of liver tumors in transgenic mice expressing high levels of hFGF19 and in mice administered a high dose of hFGF19.17,18 For that reason, in the present study, we examined FGF19 signaling at nanomolar levels.
Another issue that might lead to a discrepancy between the human and mouse systems is the difference in the structures of the respective signaling molecules (summarized in
In this report, we used in vitro model cellular systems to gain a better understanding of the activities of hFGF19 in murine in vitro and/or in vivo model systems. The results shed new light on the mechanisms underlying the experimental, pharmacological, and pathological activities of hFGF19 in a mouse model system.
Materials and Methods
Reagents
Human FGF19 was purchased from R&D systems. Heparan sulfate (HS), chondroitin sulfate (CS)-B, CS-D, and CS-E were from Seikagaku Biobusiness Corp. (Tokyo, Japan). Heparin was from Sigma (St. Louis, MO). Hepatic sGAGs were prepared as previously described. 15
Culture of BaF3 cells and DNA synthesis assay
BaF3 murine pro-B lymphoma cells were obtained from the RIKEN BioResource Center. BaF3 cells do not express detectable levels of any of the components of FGF signaling complexes (i.e., FGFR, KLB/KLA, and sGAGs). However, BaF3 cells do harbor the intracellular mediators necessary to transduce a mitogenic signal from FGFRs. BaF3 cells were stably transfected with an expression vector encoding an mFGFR subtype (mFGFR1c, mFGFR2c, mFGFR3c, or mFGFR4) with or without a vector encoding mKLB. The receptor-mediated signaling was measured based on evoked DNA synthesis, as described previously. 12 Briefly, the BaF3 transfectants were washed and plated to a density of 10 000 cells/well in 96-well plates. The cells were then stimulated with FGF19, with or without 5 µg/mL sGAG, in a total volume of 200 µL/well and incubated at 37 °C for 42 h. To verify the functionality of the FGFR signaling pathway in each transfectant, 460 pM FGF1 (R&D Systems) plus 5 µg/mL heparin was used as a positive control. The cells were labeled by adding [3H]-thymidine and were harvested 4 h later by filtration through glass fiber paper. The radioactivity incorporated into the DNA was counted in a scintillation counter.
Plasmid preparation
cDNAs encoding mFGFR1c (BC010200), mFGFR2c (X55441 and NM_010267), and mFGFR3c (BC053056) were kind gifts from Dr. David Ornitz. mFGFR4 (NM_008011) and mKLB (NM_031180.2) were cloned from mouse liver RNA. 12 cDNAs encoding hFGFR1c (NM_015850.3) and hFGFR2c (NM_000141.3) were cloned from human fetal brain RNA (Clonetech, Mountain View, CA), hFGFR3c (NM_000142.2) was from human brain RNA (Clonetech), and hFGFR4 (NM_002011.3) and hKLB (NM_175737.2) were from human liver RNA (Clonetech) as described. 15 The nucleotide sequences of these clones were confirmed to be correct.
Western blotting
Western blotting of FGFRs and KLB was performed as described previously. 12 In brief, the cells were harvested in lysis buffer supplemented with a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN), after which immunoblotting was performed using equal amounts of protein (50–100 µg), anti-FGFR1 (Santa Cruz Biotechnology, Santa Cruz, CA) and anti-mKLB (R&D Systems) primary antibodies, and horseradish peroxidase–conjugated secondary antibodies. The signals were detected using ECL Western blotting detection reagents (GE Healthcare Bio-Sciences, Piscataway Township, NJ) according to the manufacturer’s instructions.
Results
We previously showed that the BaF3 cell system originally established to determine the receptor specificity of canonical FGF signaling22–24 is also useful for studying the additional components required for endocrine FGF family signaling.12,15 Here we used the same system to expand our studies by comparing hFGF19 recognition in human and mouse receptor/co-receptor systems in the absence and presence of sGAGs. In addition to cells expressing hFGFRs and hKLB, which we prepared previously, we also prepared a series of BaF3 transfectants overexpressing one of four mouse tyrosine kinase FGFRs (mFGFR1c, mFGFR2c, mFGFR3c, or mFGFR4) with or without mKLB ( Fig. 1A ). As in the earlier studies of human systems, the intracellular kinase domains of mFGFR2c, mFGFR3c, and mFGFR4 were swapped with that of mFGFR1c to enable direct comparison of the receptor activation induced by ligand binding to the respective ectodomains. These cells were then examined for their ability to respond to hFGF19 in the presence or absence of sGAGs and mKLB. Because FGF19 transgenic mice show FGF19 concentrations of 0.9 to 3.9 nM in their peripheral blood, 18 we examined FGF19 signaling at nanomolar levels ( Fig. 1B – E ).
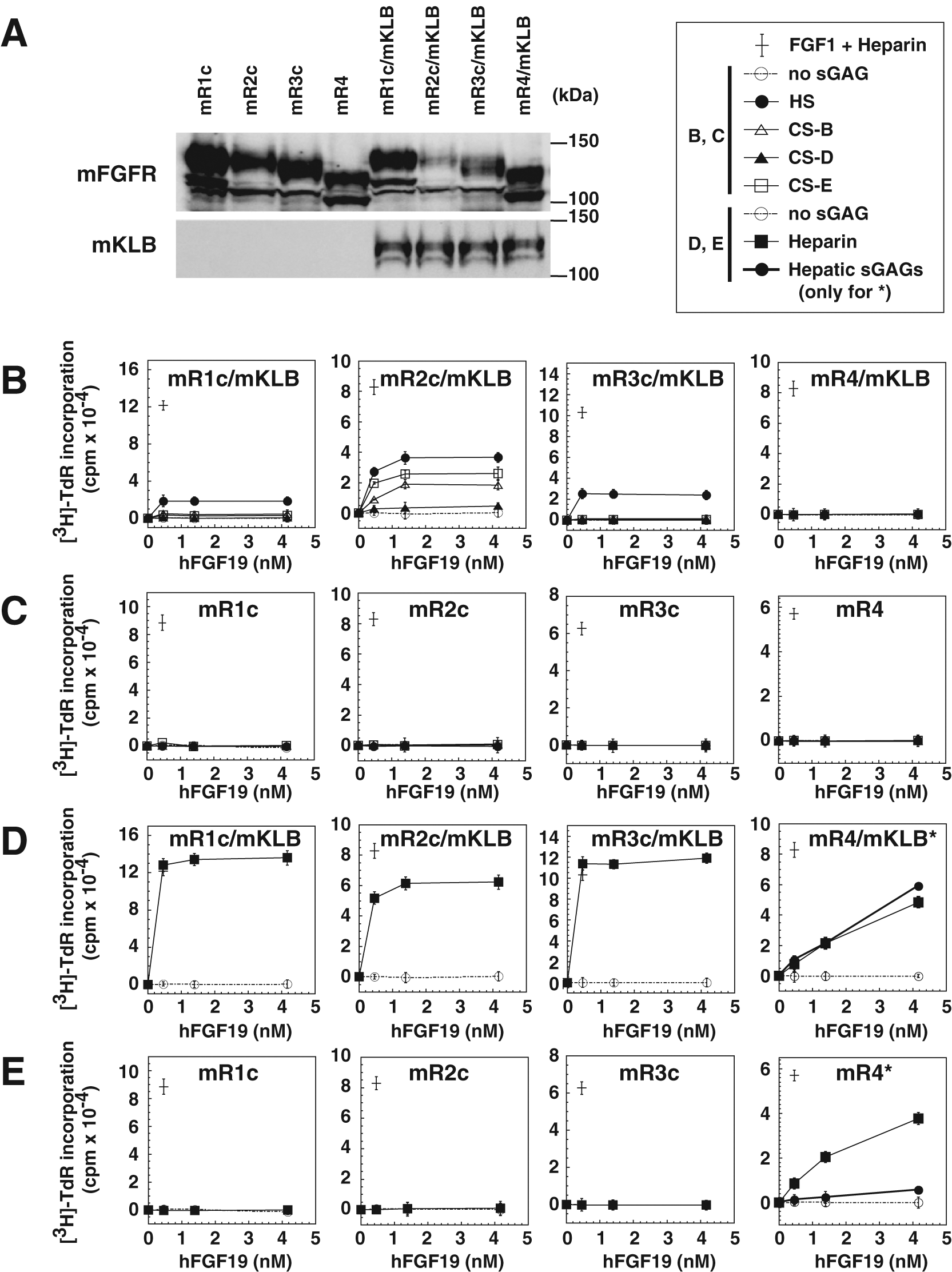
Activation of mouse (m) FGFRs by hFGF19 at nanomolar concentrations is dependent on the presence of various sGAGs and the co-expression of mKLB. (
We found that in the presence of HS and CSs, hFGF19 activated mFGFR1c, mFGFR2c, and mFGFR3c, but not mFGFR4, when co-expressed with mKLB ( Fig. 1B ). HS enhanced signaling via mFGFR1c, mFGFR2c, and mFGFR3c, whereas CS-B and CS-E enhanced signaling via mFGFR2c. Remarkably, maximal hFGF19 signals were attained at 0.4 to 1.4 nM, well below the hFGF19 concentration typically seen in transgenic mice 18 and much lower than in pharmacologic mouse experiments.19–21 These results are in stark contrast to those obtained in an hFGFR/hKLB system ( Fig. 2A ).
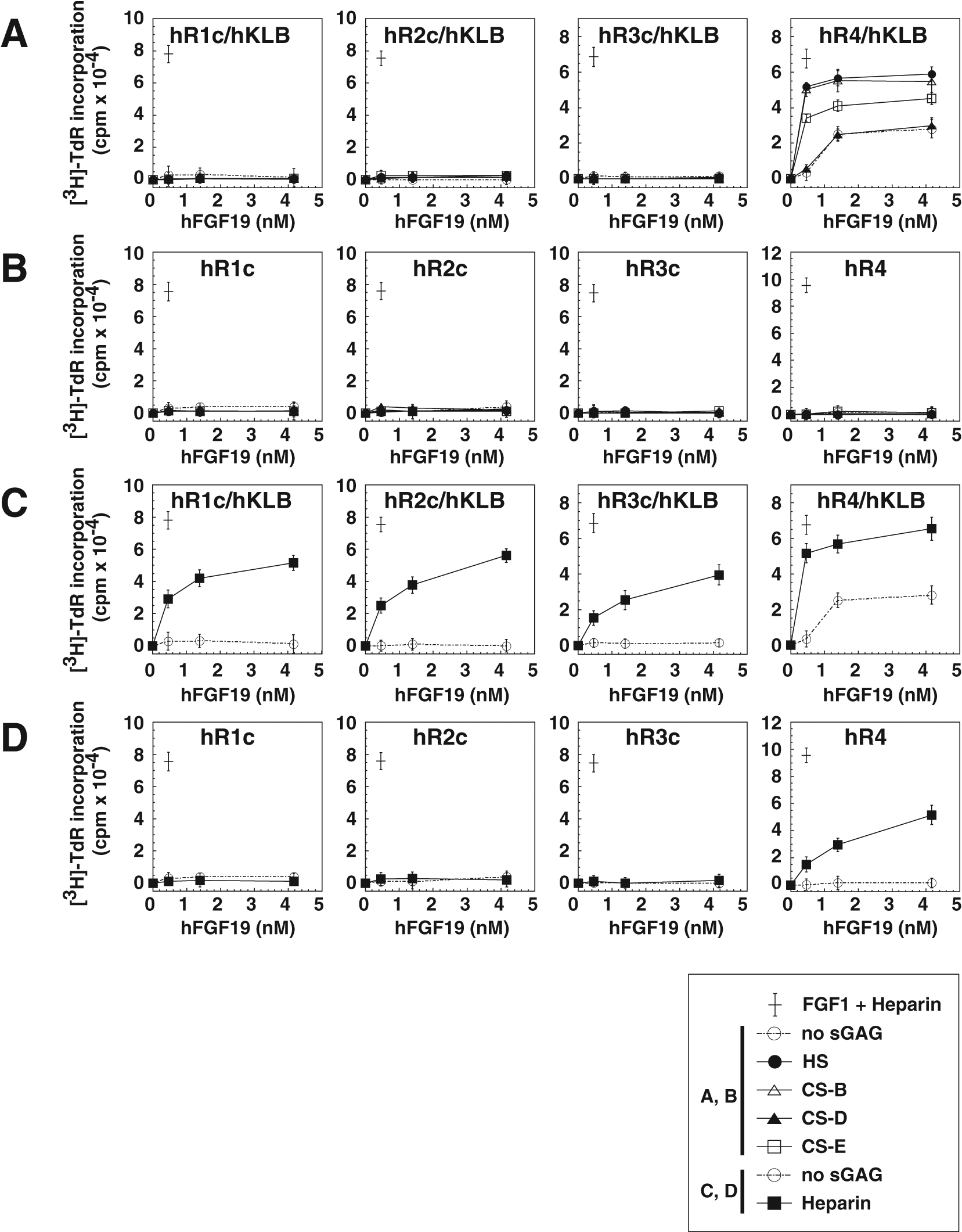
Activation of human (h) FGFRs by hFGF19 at nanomolar concentrations is dependent on the presence of various sGAGs and the co-expression of hKLB. The indicated BaF3 transfectants expressing hFGFR with/without hKLB were prepared as described previously.
15
The results shown are from experiments carried out independently of our earlier report and thus confirm the reproducibility of the results. Aside from the species of the FGFRs and KLB, the experiments were performed exactly as described in the legend to
Figure 1
. (
Using the same sGAGs and in the absence of mKLB, none of the mFGFRs were activated by hFGF19 ( Fig. 1C ). By contrast, in the presence of heparin and mKLB, hFGF19 activated all four mFGFR subtypes, as in the human system ( Fig. 1D , Fig. 2C ). Moreover, even in the absence of mKLB, heparin still enabled nanomolar hFGF19 to strongly activate mFGFR4 ( Fig. 1E ). Like heparin, hepatic sGAGs were able to strongly assist hFGF19 (0.4–1.4 nM) signaling via mFGFR4 in the presence of co-expressed mKLB ( Fig. 1D , mR4/mKLB, solid bold line) and moderately assist the signaling in the absence of co-expressed mKLB ( Fig. 1E , mR4, solid bold line). At higher concentrations (12.5 nM) of hFGF19, however, hepatic sGAGs were also able to strongly assist hFGF19 signaling via mFGFR4, even in the absence of co-expressed mKLB (described below, Fig. 3F ).
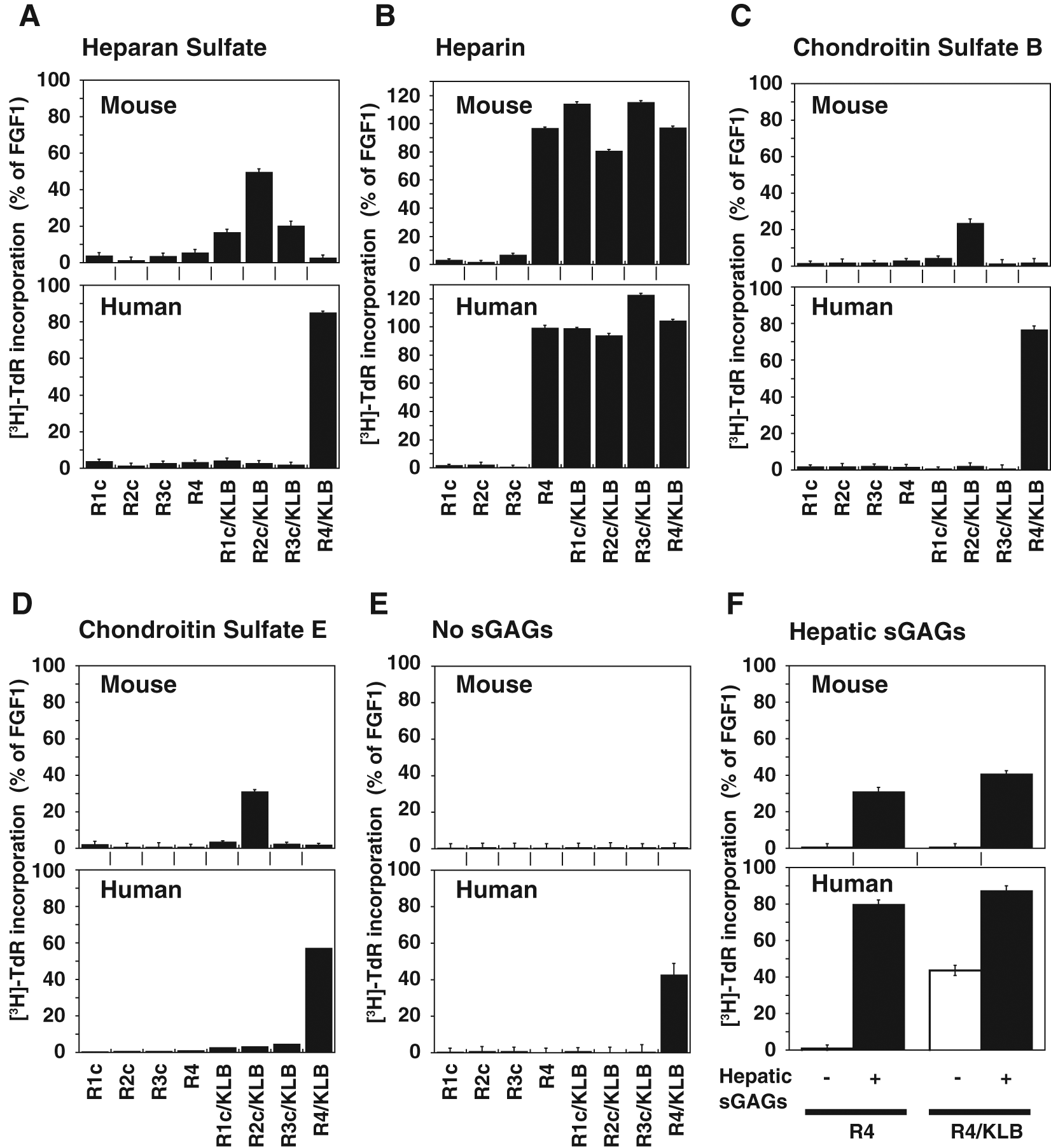
The sulfated glycosaminoglycans (sGAG)–aided receptor specificity of hFGF19 at nanomolar concentrations differs greatly between mouse and human systems. Activation of mFGFRs (the panels labeled Mouse) or hFGFRs (the panels labeled Human) by hFGF19 at 12.5 nM in the presence of sGAGs and co-expressed mKLB or hKLB, respectively, were examined. Stable BaF3 transfectants expressing mFGFR alone or mFGFR plus mKLB (
Fig. 1
) or stable BaF3 transfectants expressing hFGFR alone or hFGFR plus hKLB were stimulated with 12.5 nM hFGF19 as in
Figure 1
in the absence or presence (5 µg/mL) of the indicated GAGs. Signaling was evaluated based on evoked DNA synthesis as in
Figure 1
. Effects of heparan sulfate (
The combination of mFGFR4 and mKLB was not sufficient to mediate hFGF19 signaling in the absence of sGAGs ( Fig. 1B and D , mR4/mKLB, dotted line), indicating that sGAGs are indispensable for the mouse receptor/co-receptor system to respond to hFGF19, even in the nanomolar range. This differs from the human system, in which sGAGs were indispensable only for picomolar hFGF19 signaling via hFGFR4/hKLB. 15
We next compared the receptor signaling elicited by 12.5 nM hFGF19 in mouse and human systems in the presence of each authentic sGAG ( Fig. 3A – D ). hFGF19 has typically been studied at 100 nM in pharmacologic animal experiments19–21 or at 50 nM for cell biology studies using mouse cells and/or mouse FGFR/KLB cDNAs. 25 In the present study, the elicited signals are presented as the percentages of the signals attained with FGF1, the most efficacious FGF ligand for every FGFR subtype. The results shown for the human system ( Fig. 2 , Fig. 3 , Human) are in good agreement with our earlier findings. 15 On the other hand, when the FGFR and co-receptor KLB were of mouse origin, the target specificity of hFGF19 was quite different from that in the human system ( Fig. 3A , C – E ). In the mouse system, in the presence of HS, CS-B, or CS-E, mFGFR2c co-expressed with mKLB was most strongly activated by hFGF19, whereas mFGFR4 co-expressed with mKLB was not activated ( Fig. 3A , C , or D , respectively). By contrast, in the human system, in the presence of authentic nonheparin sGAGs (i.e., HS, CS-B, or CS-E), hFGFR4 co-expressed with hKLB was most strongly activated by hFGF19, whereas other hFGFRs co-expressed with hKLB were not activated ( Fig. 2A , Fig. 3A , C , or D ). When heparin was present, however, every subtype of mFGFR and hFGFR co-expressed with KLB from the respective species was strongly activated ( Fig. 3B ). In addition, even in the absence of KLB, heparin enabled activation of mFGFR4 and hFGFR4 by hFGF19 ( Fig. 3B ).
We also assessed the ability of hepatic sGAGs to assist signaling elicited by 12.5 nM hFGF19 via mouse and human FGFR4 in the absence and presence of the respective KLB ( Fig. 3F ). We found that in both systems, hepatic sGAG-assisted hFGF19 signaling via FGFR4 in the absence of co-expressed KLB was as strong as that in the presence of KLB. At concentrations of 3.7 nM and lower, hFGF19 signaling assisted by hepatic sGAGs in the absence of co-expressed KLB was lower than that assisted by heparin ( Fig. 1E , mR4). Thus, with an increase in the hFGF19 concentration, its KLB-independent signaling via FGFR4 becomes significant. It is noteworthy that only in the human system was there a certain level of sGAG-independent hFGF19 signaling via hFGFR4 when hKLB was co-expressed ( Fig. 2A and C , Fig. 3E ), which is consistent with our earlier report. 15
Discussion
By expressing FGFRs and KLB on the surface of cells that do not express them endogenously and supplementing sGAGs, we previously showed that sGAGs are crucial for specific and sensitive hFGF19 signaling via hFGFRs and suggested that hepatic sGAGs are involved in the highly potent and specific signaling of picomolar hFGF19 via hFGFR4 and hKLB. 15 Here we showed that the target specificity of such sGAG-assisted hFGF19 signaling in a mouse experimental system differs greatly from the physiological signaling one would expect to see in healthy humans.
It was reported that the phenotype of bile acid regulation in Fgf15 knockout mice is similar to that in Fgfr4 knockout mice, suggesting that mFGFR4 is the sole physiological receptor for mFGF15. 7 Furthermore, the chromosomal locations and structures of the genes encoding mFGF15 and hFGF19 confirmed they were orthologs.1,5,6 Both mFGF15 and hFGF19 are expressed in the intestine in response to bile acid and then secreted into the blood; in liver cells, both molecules suppress transcription of Cyp7A1, a key enzyme involved in bile acid biosynthesis.3,7 This led to an expectation that hFGF19 might exhibit similar receptor specificities in mouse and human systems. Consistent with that idea, we showed here that nanomolar hFGF19 activates mFGFR4 in the presence of heparin or hepatic sGAGs ( Fig. 1D and E ) and that the activity persisted in the absence of mKLB ( Fig. 1E ), which is similar to our findings with nanomolar hFGF19 in the human system ( Fig. 2C and D ; Fig. 3F ). However, there is a clear difference between the human and mouse systems for the other FGFR subtypes. In the presence of authentic HS or CSs, hFGF19 activated mFGFR1c, mFGFR2c, and mFGFR3c only in the presence of mKLB ( Fig. 1B ); these sGAGs did not assist hFGF19 signaling via mFGFR1c, mFGFR2c, or mFGFR3c when mKLB was absent ( Fig. 1C ). In the human system, by contrast, HS and CSs assisted hFGF19 signaling via hFGFR4 when co-expressed with hKLB but not via hFGFR1c, hFGFR2c, or hFGFR3c ( Fig. 2A ; Fig. 3A , C , and D ). Thus, the target specificity of sGAG-assisted hFGF19 signaling in the mouse system appears very different than that in the human system.
Direct assessment of mFGF15 signaling in a mouse system has not yet been achieved, either in our in vitro experiments or in an earlier in vivo study, because the activity of recombinant FGF15 protein is unstable (results not shown), as previously reported. 26 In addition, physiological concentrations of mFGF15 could not be measured. 27 To overcome these technical problems and examine the physiological function and mechanism of mFGF15, considerable effort has gone into analyzing the effects of mFGF15 knockout or infection with an mFGF15-expressing adenovirus. Through these lines of experiments, it was found that mFGF15 inhibits expression of Cyp7a1, a key enzyme involved in the synthesis of bile acid via mFGFR4 in liver. 7 It was also shown that mFGF15 suppresses neoglycogenesis in the liver by inhibiting dephosphorylation of CREB and suppression of PGC-1 via mFGFR4. 27 The possibility that mFGFR1c and mFGFR2c are involved in these responses was not addressed. In those in vivo studies, it was also not clearly demonstrated whether mKLB serves as a co-receptor for mFGF15 or whether mKLA is involved.
Our results are consistent with numerous earlier investigations in which the mouse was used to study genetic, pharmacological, and/or cell biological processes. For example, the potential utility of hFGF19 in the treatment of diabetes mellitus has been the subject of several studies.3,8,9,20 It was found that the FGFR subtype most strongly expressed in murine white adipose tissue (WAT) was mFGFR1c and that mFGFR4 expression was very low. 28 Furthermore, mFGFR1c, not mFGFR4, was responsible for hFGF19-induced phosphorylation of ERK in WAT.19,20 In addition, in vitro studies using mFGFRs and mKLB suggested mFGFR1c, mFGFR2c, and mFGFR3c are involved in regulating glucose, lipid, and energy metabolism. 3 Regulation of gallbladder filling is another hFGF19 function studied in the mouse system. In that instance, it was suggested that mFGFR3 is the receptor mediating the hFGF19-induced filling of the gallbladder in Fgfr4 knockout mice. 29
An important issue underlying the apparent difference in the receptor specificities of hFGF19 in human physiological systems and mouse experimental systems is the concentration of hFGF19 at the site of action. In many earlier experiments, hFGF19 activity was studied at very high concentrations, ranging between 10 nM and 100 nM,19–21 whereas the physiological concentration of hFGF19 in the peripheral blood of healthy humans is approximately 30 pM. 4 We previously suggested that hFGF19 at pathological concentrations may evoke aberrant signaling through various FGFRs, even in humans. 15 In mouse experiments, it was shown that 100 nM hFGF19 acts via mFGFR4 in an mKLB-independent manner to induce mouse hepatocyte proliferation. 21 We showed here that at nanomolar levels, which is the condition under which many mouse-based FGF19 studies have been conducted, hFGF19 activates mFGFR4 in the presence of heparin or hepatic sGAGs, both in the absence and presence of mKLB (Fig. 3B and F ). Mice expressing an hFGF19 transgene, which reportedly exhibit blood hFGF19 levels of 0.9 to 3.9 nM, develop hepatic tumors. 18 In the human system, activation of hFGFR4 alone reportedly transduces oncogenic signals into hepatocytes, whereas activation of FGFR4 in the presence of KLB suppresses tumorigenesis. 30 We therefore suggest that the mKLB-independent mFGFR4 activation by hFGF19 in these transgenic mice could explain the molecular basis for tumor development in those animals.
Another issue underlying the discrepancy between the human and mouse systems is the difference in the structures of the respective signaling molecules (
We also showed that authentic HS and sGAGs prepared from liver have different effects on hFGF19 receptor specificity. The hepatic sGAGs used in this study contain both HS and CSs. In cellulose acetate electrophoresis, our samples of hepatic sGAGs migrated close to the authentic HS, CS-B, and CS-E but clearly differed from heparin. 15 Heparin is a fully sulfated form of HS found in a few mammalian cells/tissues, including mast cells and intestinal mucosa. In the HS fraction of hepatic sGAGs, the content of a highly sulfated disaccharide unit [(U,6,N)S] was higher than in the authentic HS fraction but lower than in the heparin fraction. 15 It is now widely accepted that, for canonical FGFs, HS forms a signaling complex with the corresponding FGFR. In those instances, sulfate groups at specific positions play crucial roles in the interaction of heparin/HS with the respective FGF ligand and FGFR. For example, 6-O-sulfation of HS differentially regulates various FGF-dependent signals. 33 We therefore suggest that hepatic sGAGs contribute to the formation of active signaling complexes composed of hFGF19, FGFR4, and KLB in liver. Hepatic sGAGs assisted activation of mFGFR4 by hFGF19 in a manner similar to heparin ( Fig. 1D , mR4/mKLB; Fig. 3F ). Given the tissue-dependent structural heterogeneity of HS, 34 we suggest that in the liver, endogenous HS may act in a manner similar to heparin with respect to activation of mFGFR4. In other mouse tissues, by contrast, where the pattern of endogenous HS sulfation may be different than in liver, endogenous HS might assist mKLB-dependent, nanomolar hFGF19-induced activation of mFGFR1c, mFGFR2c, and mFGFR3c.
Considering that numerous studies on hFGF19 have been conducted in mouse systems in an effort to develop drugs for humans, our results strongly suggest that the function and mechanism of hFGF19 signaling identified using mouse systems should be reevaluated.
Footnotes
Acknowledgements
We deeply thank Dr. Yukishige Ito for kindly providing access to his laboratory facilities at RIKEN; Professors Yukio Ishimi and Zheng Yu Wang for tutorial assistance; and Mss. Miho Kimura-Ueki, Emi Honda, Akiko Komi-Kuramochi, and Junko Oki at AIST for providing materials and technical help. This article was edited by Dr. William Goldman.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: M.N. was supported by an Innovation School Grant from AIST. This research was supported in part by the research budget of AIST and by the Aid Of Suffering Researchers budget of RIKEN.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.