
Abstract
Chikungunya virus (CHIKV) is a mosquito-transmitted pathogen responsible for an acute infection of abrupt onset, characterized by high fever, polyarthralgia, myalgia, headaches, chills, and rash. In 2006, CHIKV was responsible for an epidemic outbreak of unprecedented magnitude in the Indian Ocean, stressing the need for therapeutic approaches. Since then, we have acquired a better understanding of CHIKV biology, but we are still missing active molecules against this reemerging pathogen. We recently reported that the nonstructural nsP2 protein of CHIKV induces a transcriptional shutoff that allows the virus to block cellular antiviral response. This was demonstrated using various luciferase-based reporter gene assays, including a trans-reporter system where Gal4 DNA binding domain is fused to Fos transcription factor. Here, we turned this assay into a high-throughput screening system to identify small molecules targeting nsP2-mediated shutoff. Among 3040 molecules tested, we identified one natural compound that partially blocks nsP2 activity and inhibits CHIKV replication in vitro. This proof of concept suggests that similar functional assays could be developed to target other viral proteins mediating a cellular shutoff and identify innovative therapeutic molecules.
Introduction
Luciferase-based reporter assays are efficient tools to monitor biological processes such as gene transcription, cellular viability, or protein-protein interactions.1–3 In the virology field, recombinant viruses have been engineered by reverse genetics to express luciferase from additional transcription units or fused to viral proteins.4–8 Artificial minigenomes that express luciferase in the presence of viral replication components have also been developed.9,10 Although these systems provide convenient ways to monitor the activity of viral replication machinery in culture cells, they show some limitations when screening chemical libraries for antiviral molecules. Indeed, such systems do not discriminate between toxic and antiviral molecules since both decrease luciferase expression in culture wells. As a consequence, toxic molecules must be filtered out with a counterscreen, thus adding one more step to the screening pipeline. This is true when using luciferase or any reporter gene to monitor viral replication. This motivated the search for alternative systems where potential antiviral molecules are identified based on enhanced, rather than decreased, luciferase expression. Interestingly, viruses express essential virulence factors that, among various functions, often promote viral replication by blocking specific cellular processes such as innate immunity, gene transcription, or cap-dependent protein translation. In theory, chemical compounds that interfere with such virulence factors, and thus prevent virus interactions with targeted cellular functions, should exhibit antiviral activities as demonstrated for Nef of the human immunodeficiency virus (HIV). 11 Furthermore, reporter assays can be designed so that luciferase activity is induced when cellular functions are properly restored, thus providing a way to screen chemical libraries for inhibitors of viral virulence factors. In this report, we established a proof of concept using Chikungunya virus (CHIKV) as a model.
CHIKV is an enveloped virus with a positive, single-strand RNA genome. It belongs to the Togaviridae family from the alphavirus genus and is transmitted by Aedes mosquito bites through either urban or sylvatic transmission cycles. CHIKV is endemic in Africa, India, and Southeast Asia but was responsible for a major epidemic across the Indian Ocean in 2005–2006. 12 This CHIKV outbreak was accompanied by more severe symptoms than previously reported, including complications and deaths, and the development of chronic arthralgia up to years after infection. In the absence of specific therapy, treatment was only symptomatic, combining analgesic and anti-inflammatory agents. Since then, CHIKV also has become a threat in southern Europe since dozens of cases have been reported both in Italy and in the south of France.13,14 Altogether, this calls for the development of both vaccine and therapeutic strategies to protect human populations from this reemerging pathogen.12,15
Recently, the passive transfer of immune serum was shown to protect mice from a CHIKV challenge, suggesting that antibody-based therapies could be useful.16–18 However, this approach is not cost-effective and easily scalable so that alternative strategies must be developed. In a wild-type mouse model, interferon (IFN)–α treatment was shown to prevent CHIKV-induced symptoms but only when injected before, but not after, infection. 18 Ribavirin was also tested on a small cohort of human patients with chronic CHIKV arthralgia. 19 This broad-spectrum antiviral molecule showed some but limited beneficial effects on patient symptoms, in agreement with in vitro experiments showing a weak antiviral activity. 20 Chloroquine, an alkalinizing agent that blocks viral entry and maturation of CHIKV particles, 21 was also evaluated in vivo. Despite some measurable effect on CHIKV-induced chronic arthralgia, 22 this molecule had no beneficial effect during the acute phase of the disease. 23 More recently, it was shown that mycophenolic acid, a potent inhibitor of guanine nucleotide biosynthesis, efficiently blocked CHIKV replication in vitro. 24 Few other molecules, including 5,7-dihydroxyflavones, were recently characterized as CHIKV inhibitors by using phenotypic assays based on CHIKV replicons or recombinant viruses expressing reporter genes. Whether these different molecules represent leads for the development, an in vivo therapy remains to be determined. 10 Altogether, this demonstrates the lack of efficient, cost-effective antiviral therapy against CHIKV infection.
CHIKV genome is an 11.8-kb RNA molecule with a 5′ cap allowing its direct translation into a full-length nonstructural polyprotein (nsP) called P1234.25,26 This precursor progressively cleaves itself to produce nsP1, nsP2, nsP3, and nsP4. These proteins assemble into a RNA polymerase complex that replicates viral genome. This enzymatic complex also transcribes a 26S subgenomic RNA from the 3′ extremity of the viral genome. This messenger RNA is translated into a polyprotein precursor, which is cleaved by a combination of viral and cellular enzymes to produce mature structural proteins C, E3, E2, 6k, and E1 before assembly into viral particles. 27 The nonstructural protein nsP2 is particularly interesting because of its multiple enzymatic activities and functional roles. This viral protein exhibits an N-terminal domain with an RNA and nucleotide triphosphatase (NTPase) activity, which is involved in viral RNA capping but also fuels with energy the RNA helicase domain of nsP2. At the C-terminus, nsP2 presents a protease domain, which is responsible for the autocatalytic cleavage of the nonstructural polyprotein, and a methyltransferase-like region of unknown function. In addition to its different roles as cofactors of the viral polymerase complex, CHIKV nsP2 is a virulence factor. In particular, this viral protein has been shown to inhibit type I/II interferon-stimulated JAK/STAT signaling and to block cellular gene transcription (“transcriptional shutoff”) by inducing the degradation of the RNA polymerase II subunit Rpb1.28–31 These mechanisms allow the virus to blunt the host antiviral response and defense mechanisms.
Altogether, these different activities and functions make nsP2 a potential target for CHIKV inhibitors. In a recent report where we characterized the transcriptional shutoff induced by CHIKV nsP2 in human cells, 30 we showed the potent inhibition of a trans-reporter assay where luciferase gene expression is induced by an artificial transcription factor called Gal4-DB-Fos. Here we used this transcriptional assay as a screening test to identify small molecules that alleviate the transcriptional shutoff induced by this viral protein.
Material and Methods
Chemical Compound Library
The chemical library from the Institut de Chimie des Substances Naturelles (ICSN, Gif-sur-Yvette, France) is currently composed of 4640 natural products, molecules derived from natural substances, and synthetic compounds (average molecular weight [MW] of 400 ± 200). Of each compound, 1 mg was dissolved in 1 mL of DMSO and arrayed in a 96-well format. Compound ID1452-2 is a synthetic intermediate that has not been described so far. Nuclear magnetic resonance (NMR) data (1H and 13C) and mass spectrometry (MS) analysis (data not shown) all support the structure depicted in Figure 1 .
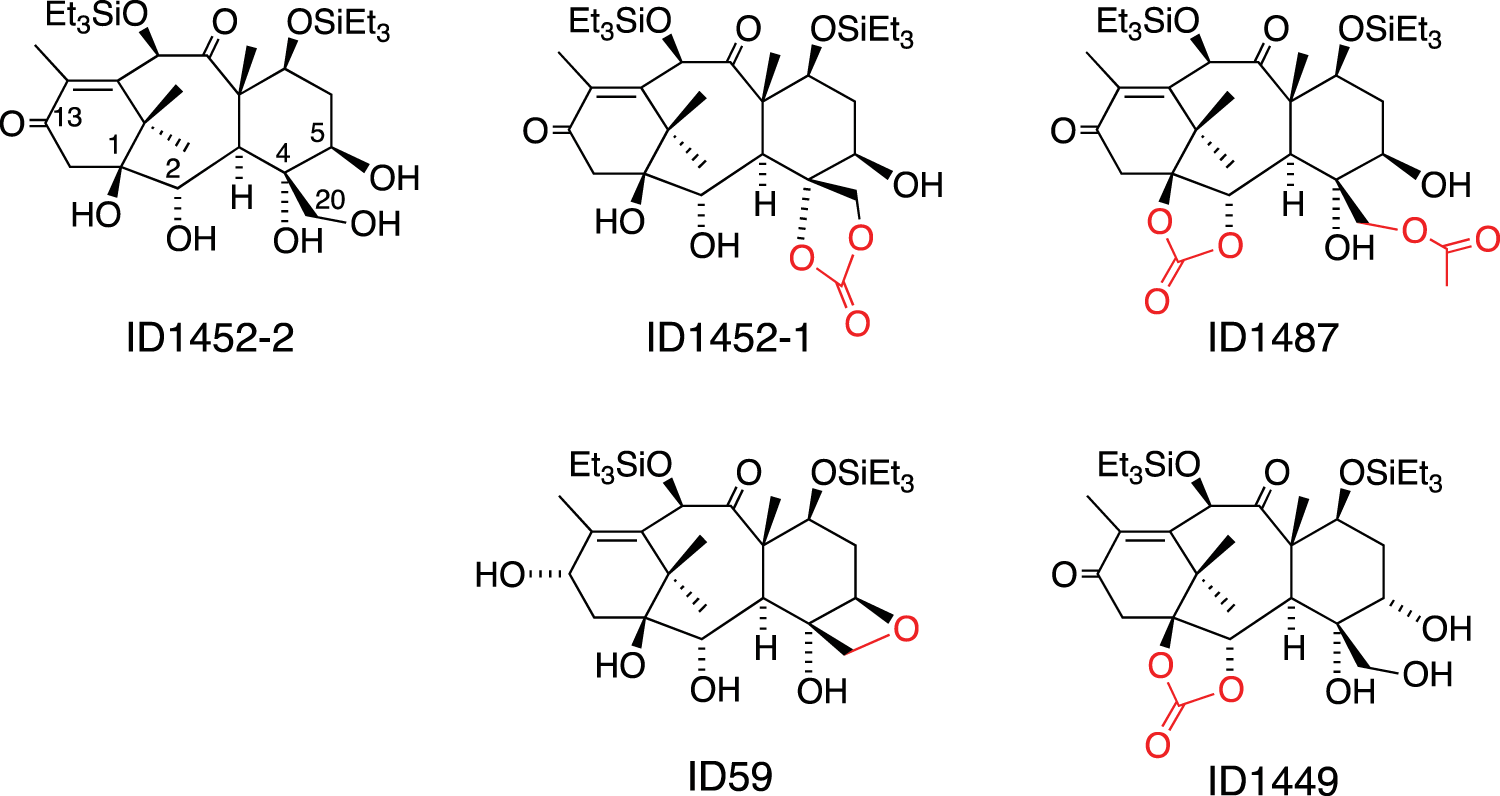
Structures of ID1452-2 and four analogues present in the Institut de Chimie des Substances Naturelles (ICSN) library. Differences between ID1452-2 and analogues are shown in red.
Cell Lines and Viruses
HEK-293T cells (ATCC, Manassas, VA) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-Invitrogen, Carlsbad, CA) containing 10% fetal calf serum (FCS), penicillin, and streptomycin at 37 °C and 5% CO2. Cell viability was determined by quantitation of adenosine triphosphate (ATP) in culture wells using the CellTiter-Glo Assay (Promega, Madison, WI) following the manufacturer’s recommendations. The recombinant CHIKV strain expressing Renilla luciferase from a coding sequence inserted between nsP3 and nsP4 nonstructural proteins has already been described.30,32 Virus stock was produced on VERO cells and titrated by TCID50 on HEK-293T cells. The recombinant measles virus strain expressing firefly luciferase from an additional transcription unit has been previously described (rMV2/Luc). 8 Virus stock was produced on VERO cells and titrated by TCID50 on VERO cells.
Luciferase Reporter Gene Assay
The ORF encoding for nsP2 from CHIKV was transferred by in vitro recombination (LR cloning reaction, Gateway technology, Invitrogen) from pDONR207 into the pCI-neo-3xFLAG vector to be expressed in human cells in fusion with a 3xFLAG tag. 30 HEK-293T cells were co-transfected with pGal4-UAS-Luc reporter plasmid (75 ng/well; kindly provided by Dr. Yves Jacob, Institut Pasteur, Paris, France) and pM vector (75 ng/well; Clontech, Mountain View, CA) expressing Gal4-BD fused to Fos, together with pCI-neo-3xFLAG vector, either empty or expressing CHIKV nsP2 (75 ng/well). Cell transfection was performed with a 1-mg/mL polyethylenimine (PEI “Max”; Polysciences, Warrington, PA) solution adjusted at pH 7. For 100 culture wells, 70 µL PEI solution was diluted in 500 µL DMEM without FCS. In parallel, 7.5 µg of each one of the plasmids required for the assay was mixed into 500 µL DMEM without FCS. PEI and plasmids were mixed together with a vortex, and transfection mix was incubated for 30 min at room temperature. Finally, 2.5 × 106 cells were added to the mix together with 9 mL DMEM containing FCS and antibiotics. After 24 h of culture, firefly luciferase activity was determined using the Bright-Glo reagent following the manufacturer’s recommendations (Promega).
Screening Procedure
All robotic steps were performed on a TECAN Freedom EVO platform (TECAN, Männedorf, Switzerland). Compounds were transferred from mother plates into white, flat bottom, bar-coded tissue culture 96-well plates (Greiner Bio One, Monroe, NC): 1 µL of a DMSO solution at 1 mg/mL was spiked into dry wells of daughter plates (80 compounds per plate). Human HEK-293T cells were co-transfected with the three plasmids as described above and seeded into compound-containing daughter plates. Then, 2.5 × 104 cells resuspended in 100 µL culture medium were dispensed in each well. For each plate, columns 1 and 12 were dedicated to controls. The first column was spiked with 1 µL DMSO alone (positive control with the maximal rate of nsP2 inhibition). The last column was devoted to negative controls with cells co-transfected with the luciferase reporter plasmid, the Gal4-BD-Fos expression plasmid, and the empty pCI-neo-3xFLAG vector (no nsP2 expression). After 24 h of incubation at 37 °C in the presence of 5% CO2, the firefly luciferase substrate (Bright-Glo; Promega) was added directly into the wells (50 µL) and luciferase activity was measured 6 min later on a Safire2TM (TECAN) using a 100-ms integration time.
Viral Infections
HEK-293T cells were dispensed in a microplate at 3 × 104 cells/well in 100 µL culture medium. ID1452-2 was added to the wells at final concentrations of 7.75, 15.5, or 31 µM (5, 10, or 20 µg/mL, respectively). Finally, cells were infected with CHIKV-Luc (multiplicity of infection [MOI] = 0.2) or rMV2/Luc (MOI = 1). After 24 h of culture, CHIKV-Luc and rMV2/Luc replication was revealed with the Renilla-Glo and Bright-Glo reagents (Promega), respectively. This time point corresponds to the exponential phase of viral replication, before Renilla or Luciferase activities reach a plateau and viruses induce cytopathic effects and cell death. Cell viability in noninfected culture wells treated with DMSO alone or ID1452-2 was determined by quantitation of ATP in culture wells using the CellTiter-Glo Assay (Promega) following the manufacturer’s recommendations.
Results
Assay Development and Optimization for High-Throughput Screening
We recently demonstrated that CHIKV nsP2 induces a transcriptional shutoff when expressed in human cells as assessed by the inhibition of various reporter gene constructs. In particular, this was shown when using a luciferase reporter gene of which expression is under the control of Fos, a potent transcription factor of human origin. In this cell-based functional assay ( Fig. 2 ), human Fos is fused to the DNA-binding domain of Gal4 (Gal4-BD), a modular transcription factor isolated from yeast. This hybrid transcription factor efficiently binds to Gal4-UAS sites located within the promoter region of a luciferase reporter gene of an episomal plasmid and promotes its expression. When CHIKV nsP2 is coexpressed, Fos-induced expression of luciferase is strongly inhibited. Here, we considered this functional assay to identify inhibitors of CHIKV nsP2 that restore luciferase expression and thus alleviate the transcriptional shutoff induced by this viral protein.
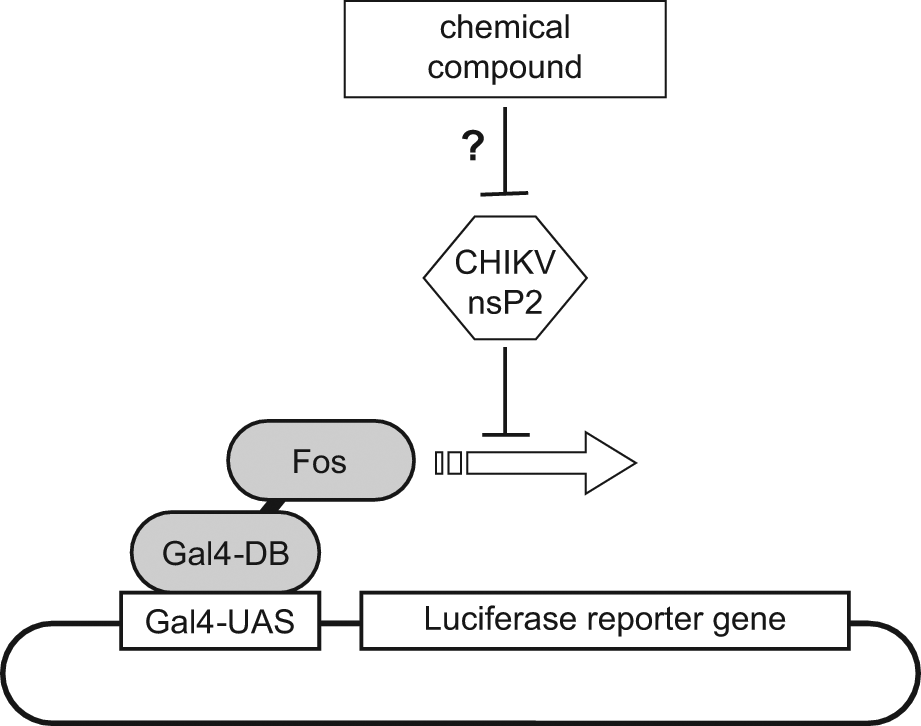
Schematic of the screening assay. Luciferase reporter gene expression is dependent on Fos transcription factor. In the presence of CHIKV nsP2, luciferase expression is inhibited. Our goal was to identify chemical compounds that alleviate nsP2-mediated shutoff on gene transcription and thus restore luciferase expression.
Cell transfection and culture conditions were determined to perform the assay in a high-throughput, 96-well format. To reduce experimental cost, transfection was performed with bulk PEI. Optimal plasmid to PEI ratio was first determined using a reporter plasmid encoding for the red fluorescent protein Cherry and was set at 1:3 (w/w) for 2.5 × 104 cells per well (see Materials and Methods). These conditions were used to co-transfect human HEK-293T cells with all three plasmids from the assay (i.e., the luciferase reporter plasmid and expression plasmids encoding for Gal4-BD-Fos and CHIKV nsP2). Cells were dispensed into 96-well white plastic culture plates suitable for bioluminescence reading. After 24 h of culture, luciferase activity was determined by adding the luciferase substrate directly into the wells.
We then evaluated this protocol on a few microplates before running the screen. To perform this analysis in relevant conditions, DMSO was dispensed in all culture wells since all compounds from the chemical library were diluted in this solvent. All screening plates were organized along the same pattern, with first and last columns containing positive and negative controls, respectively. Positive controls correspond to cells co-transfected with all three plasmids in the presence of DMSO alone. Negative controls are similar, but cell transfection was performed in the absence of CHIKV nsP2. In such experimental settings, a 14-fold (±4) reduction of luciferase activity was observed between negative and positive controls. This result is in agreement with our recent report 30 as luciferase inhibition was even more pronounced than previously observed in low-throughput settings. Furthermore, the Z′ factor, 33 which was calculated using internal control wells, was above 0.5 across all tested plates. Altogether, this demonstrated that our assay is robust, reliable, and discriminant enough to isolate nsP2 inhibitors.
Assay Validation
The screening campaign was carried out on a chemical library of 3040 compounds originating from the ICSN. The library was arrayed in 96-well microplates as single compounds at a constant 1:100 dilution (1% final DMSO concentration, compatible with cell viability) with a final concentration of 10 µg/mL (average concentration of 30 ± 13 µM). For each plate, data were normalized and expressed as percentage of inhibition relief relative to positive and negative controls using the following formula: % inhibition relief = (luciferase activity for a given compound – mean of controls with nsP2)/(mean of controls without nsP2 – mean of controls with nsP2) × 100. This allowed us to level potential day-to-day variations inherent to transfection efficiency. Z′ value ( Fig. 3A ), coefficient of variation (CV), and signal-to-background (S/B) ratio were determined to be 0.786 ± 0.074 (no value below 0.5), 6.1% ± 2.3%, and 13.9 ± 4.4, respectively, thus demonstrating the robustness of our assay, which can be categorized as excellent. 33 Hits were defined as compounds exhibiting more than 10% of inhibition relief ( Fig. 3B , example of a plate with a hit). Five compounds were selected based on this threshold. To further validate and confirm hits selected from the screen, we have retested their activity in dose-response experiments. In parallel, we have checked for their cellular toxicity using the CellTiter-Glo assay (Promega). ID1452-2 was the only molecule that (1) did not show any substantial toxicity at concentrations below 31 µM (20 µg/mL) ( Fig. 4A ) and (2) inhibited nsP2 effects on luciferase expression in a dose-dependent and reproducible manner ( Fig. 4B ). The ID1452-2 structure is depicted in Figure 1 . This molecule was then tested for its potential impact on CHIKV replication in vitro.
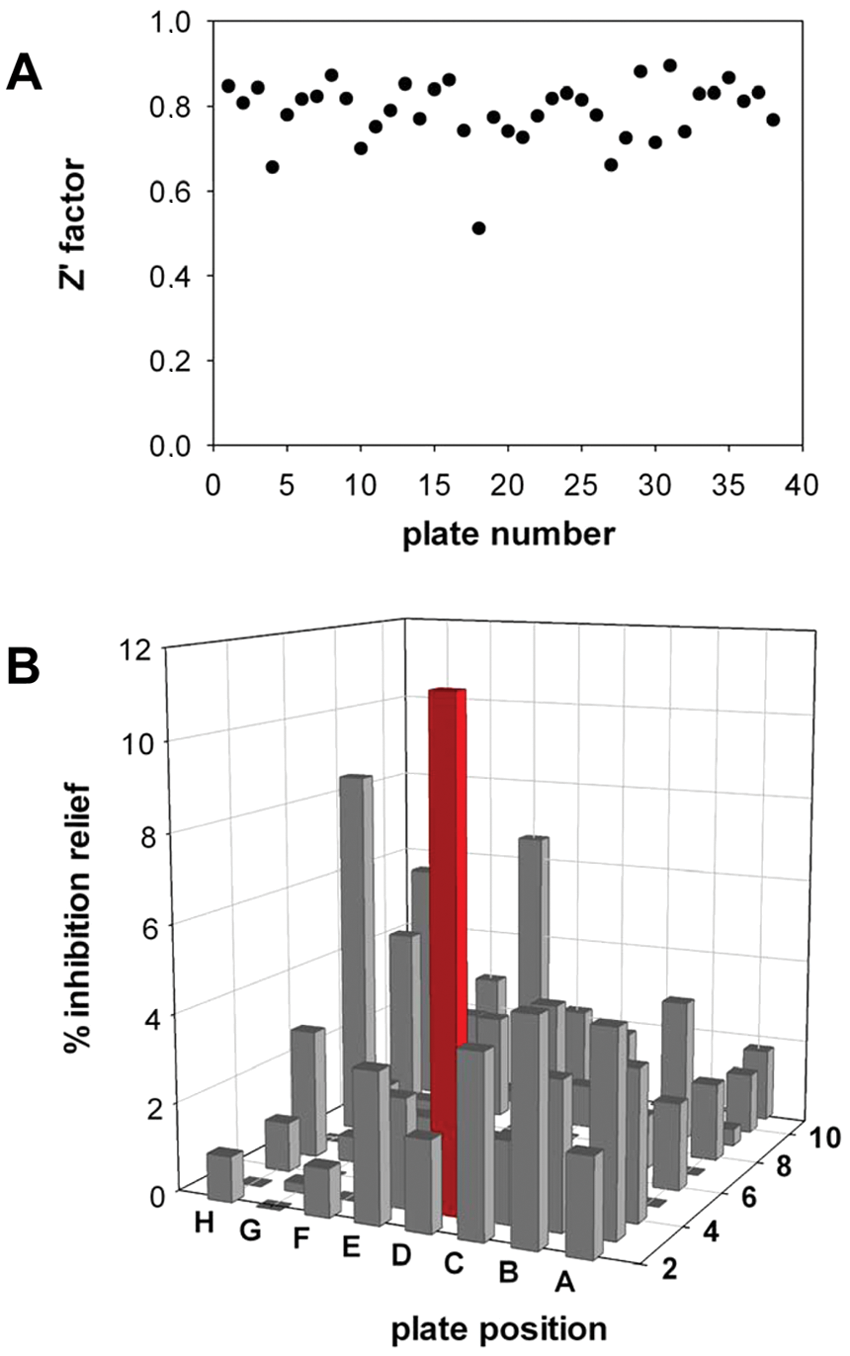
Pilot screening campaign. (
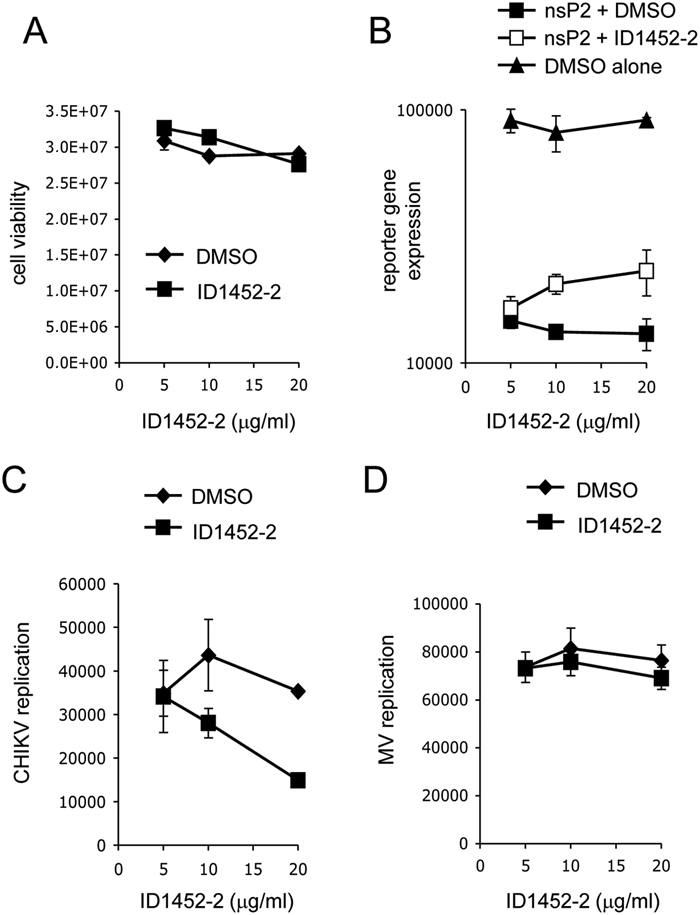
Partial relief of nsP2-mediated shutoff and inhibition of Chikungunya virus (CHIKV) replication by ID1452-2. (
To evaluate CHIKV replication in human cell cultures, we used a recombinant virus expressing the Renilla luciferase as a reporter of viral genes expression (CHIKV-Luc). 38 HEK-293T cells were infected with CHIKV-Luc (MOI = 0.2) in the presence of ID1452-2 at 7.75, 15.5, or 31 µM (5, 10, or 20 µg/mL, respectively). At 24 h postinfection, Renilla luciferase activity was measured to evaluate CHIKV replication. As shown in Figure 4C , a 50% reduction of Renilla luciferase activity was observed in the presence of ID1452-2 at 31 µM, thus demonstrating a weak but statistically significant inhibition of viral replication (IC50 = 31 µM). As a control, the same experiment was performed using a recombinant measles virus expressing the firefly luciferase as a reporter (MV-Luc). MV is a negative-strand RNA virus that is completely unrelated to CHIKV and does not express any protein related to nsP2. As shown in Figure 4D , ID1452-2 did not affect MV replication in vitro, supporting the specificity CHIKV inhibition by ID1452-2. Altogether, these results demonstrate that our screening assay can be used to isolate CHIKV inhibitors that target nsP2-induced cellular shutoff.
Discussion
In the current report, we describe a phenotypic assay to select for inhibitors of CHIKV that specifically target the cellular shutoff induced by nsP2. Among 3040 molecules tested, we identified ID1452-2, a natural compound derivative, which partially blocks nsP2 activity and inhibits CHIKV replication in vitro, making the proof of concept that a functional assay targeting virulence factors that mediate cellular shutoff allows the identification of potential therapeutic molecules. This assay was originally developed because CHIKV handling requires a biosafety level 3 (BSL-3) containment, which critically hinders the screening procedure when looking for antiviral compounds. Pohjala et al. 10 recently described a BKH-based cell line containing a stable CHIKV replicon to solve this issue. In this system, structural proteins were deleted from the CHIKV genome and replaced by a synthetic sequence encoding for puromycin acetyltransferase in a fusion with enhanced green fluorescent protein (EGFP), thus providing both selection and screening systems. The Renilla luciferase reporter was also inserted within the nsP3 sequence of their CHIKV replicon to achieve high-throughput quantitative screens. Finally, the stable expression of this replicon in BHK cells was required to introduce several mutations within the C-terminal domain of nsP2 in order to prevent the induction of a cellular shutoff that was highly toxic for transfected cells. This allowed Pohjala et al. to identify several inhibitors of CHIKV with IC50s in the low millimolar (100 µM) range. 10 Although powerful, this assay is not suitable to identify inhibitors of viral entry or nsP2-induced cellular shutoff, two potential targets for antiviral drugs against CHIKV infection. Furthermore, their assay did not readily discriminate between antiviral and toxic molecules that both induce a decrease in the Renilla luciferase signal. In the future, viral entry could be easily monitored in a BSL-2 containment laboratory by using pseudotyped lentiviral vectors that assemble into virus-like particles. 16 The screening assay developed in our laboratory monitors the cellular shutoff induced by CHIKV nsP2 in a BSL-2 containment.
CHIKV, as previously described for other Old World alphaviruses, induces a cellular shutoff at both transcriptional and translational levels and blocks interferon-mediated signaling.28–30 This prevents the induction of an antiviral state within infected cells, thus promoting viral replication.34,35 It has been established that nsP2 is critically involved in this process.28,30 Although the helicase and protease domains are dispensable, the methyltransferase-like domain at the C-terminus of nsP2 is essential.28,30 Furthermore, CHIKV replicons carrying a mutation in this domain (P718G) were severely impaired for viral replication, thus establishing a clear correlation with nsP2 activity as a virulence factor. This suggested that nsP2-mediated shutoff and inhibition of interferon signaling is a potential target for antiviral drugs. Results presented in this report support this notion since ID1452-2, a chemical compound that altered nsP2-induced shutoff, also impaired CHIKV replication in vitro.
ID1452-2 is a synthetic intermediate obtained during structure-activity relationship studies on the taxoid oxetan ring. The ICSN chemical library contains other compounds structurally close to this derivative ( Fig. 1 ). These compounds showed barely detectable or no activity in this assay. These results are too preliminary to really determine the important features in ID1452-2, but they show that our assay is quite selective and can discriminate between very close structures. It must be emphasized that the activity of ID1452-2 is quite limited, and the hit rate of our assay was low. This could be explained by the fact that our phenotypic assay combines several stringent criteria, including membrane permeability to compounds, cell viability, functional gene expression, and inhibition of nsP2 activity. Nevertheless, our study demonstrates that our assay enables identification of suitable molecules. This suggests that more active molecules could be identified by screening different and/or larger chemical libraries.
Fighting viruses requires the development of both vaccine and therapeutic strategies to confront all epidemic situations. Our current arsenal against viruses is quite limited and includes few viral polymerase inhibitors that cripple viral replication and recombinant interferons to boost the host antiviral system. More recently, protease inhibitors have been developed with success against HIV and hepatitis C virus and are developed against other viruses. 36 However, both polymerase and protease inhibitors target enzymatic activities carried by viral proteins. In the near future, a new class of antiviral molecules should emerge that will aim at blocking interactions between viral and cellular components. Indeed, substantial progress has been made in our understanding of virus-host protein interactions that are essential to viral replication. For example, new drugs that are currently tested in the clinic specifically target the interaction between HIV integrase and the cellular factor LEDGF. 37 Among viral proteins, virulence factors that are involved in the negative control of antiviral responses are highly connected to the host proteome and thus represent potential targets for antiviral strategies. In the present report, we show that nsP2 from CHIKV, which interacts with numerous cellular proteins to control gene transcription and interferon signaling, can be targeted. Whether ID1452-2 interferes with nsP2 binding to its partners will require further investigations. However, the current report illustrates the potential of phenotypic assays designed in a way to identify chemical compounds that target virulence factors.
Footnotes
Acknowledgements
We thank Dr Y. Jacob for technical advices, fruitful discussion, and the kind gift of reagents.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Programme Interdisciplinaire—CNRS—Maladies Infectieuses Emergentes, the Institut Pasteur through the Programme Transversal de Recherche (PTR No. 201: Chikungunya), the Centre National de la Recherche Scientifique (CNRS), the Institut National de la Santé Et de la Recherche Médicale (INSERM), and the Conseil Régional d’Ile-de-France (Chemical Library Project, grants nos. I 06-222/R and I 09-1739/R to H.M.-L.). A. L. was the recipient of a postdoctoral fellowship from the Conseil Régional d’Ile-de-France.