
Abstract
Alternative splicing (AS) is an efficient mechanism that involves the generation of transcriptome and protein diversity from a single gene. Defects in pre–messenger RNA (mRNA) splicing are an important cause of numerous diseases, including cancer. AS of pre-mRNA as a target for cancer therapy has not been well studied. We have reported previously that a splicing factor, polypyrimidine tract-binding protein (PTB), is overexpressed in ovarian tumors compared with matched normal controls, and knockdown of PTB expression by short-hairpin RNA impairs ovarian tumor cell growth, colony formation, and invasiveness. Given the complexity of PTB’s molecular functions, a chemical method for controlling PTB activity might provide a therapeutic and experimental tool. However, no commercially available PTB inhibitors have yet been described. To expand our ability to find novel inhibitors, we developed a robust, fluorometric, cell-based high-throughput screening assay in 96-well plates that reports on the splicing activity of PTB. In an attempt to use the cells for large-scale chemical screens to identify PTB modulators, we established cell lines stably expressing the reporter gene. Our results suggest that this high-throughput assay could be used to identify small-molecule modulators of PTB activity. Based on these findings and the role that upregulated PTB has on cell proliferation and malignant properties of tumors, targeting PTB for inhibition with small molecules offers a promising strategy for cancer therapy.
Introduction
Pre–messenger RNA (pre-mRNA) splicing is one of the essential steps in the maturation of eukaryotic genes, whereas alternative splicing is an efficient mechanism that involves generation of transcriptome and proteomic diversity from a single gene. 1 Defects in mRNA splicing are an important cause of disease. 2 At least 15% of all disease-causing single base-pair (bp) mutations affect splicing 3 ; therefore, the splicing process and its regulation are of great interest. Many genes associated with tumorigenesis, such as oncogenes, tumor suppressor genes, cell cycle–related genes, and apoptosis-related genes, undergo alternative splicing. 4 In tumors, aberrant splicing usually arises from variations in the relative amounts/activity of regulatory splicing factors. 4 Although a definitive causal relationship between AS and cancer remains to be illuminated, it is clear that the expression of specific splice variants of many cancer-related genes can directly contribute to the oncogenic phenotype and has a determinative role in many aspects of tumorigenesis and in the development of resistance to treatments. 5
In many cases, altered splicing patterns in cancer are very likely due to changes in trans-acting splicing factors rather than cis-elements. Two highly conserved trans-acting protein families, SR proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs), are essential factors required for alternative splicing. 6 SR proteins usually bind to splicing enhancers and activate splicing at nearby splice sites. 7 In contrast, hnRNPs usually bind to splicing silencers and antagonize the activity of SR proteins in a concentration-dependent manner.7,8
One of the most intensively studied regulators of alternative splicing is the polypyrimidine tract-binding protein (PTB, also known as hnRNP I or PTBP1). 9 In addition to its pre-mRNA splicing activity, PTB is also found in the cytoplasm, where it has roles in the transportation and stabilization of mRNA. 10 It is also recruited to stimulate the internal ribosome binding sites (IRES)–mediated translation initiation from cellular and poliovirus mRNAs. 11 PTB is a ubiquitously expressed protein, but its levels among different tissues and cells vary substantially. 12 We13,14 and others 15 have found that PTB is overexpressed in human epithelial ovarian tumors and glioblastomas, respectively, compared with normal tissues. Moreover, we have shown that PTB overexpression is associated with tumor malignancy, and knockdown of PTB expression by short-hairpin RNA (shRNA) impaired ovarian tumor cell growth and malignant properties in vitro. 14 Several working models have been proposed for PTB activity, including binding to the polypyrimidine tract to looping out of repressed exons or multimeric assembly across exons to mask splice sites.9,16
The targeting of aberrant splicing is an emerging strategy to combat certain types of cancers. Some methods use direct targeting of alternatively spliced protein isoforms.17,18 Recent studies focused on other methods that alter the alternative splicing process itself as a therapeutic intervention in cancer.19,20 The key difference in these methods is whether small molecules target transcripts directly or work by affecting trans-acting splicing factors. We have focused our efforts on designing a versatile cell-based assay to screen large numbers of small-molecule compounds for their effects on a trans-acting splicing factor, PTB. However, finding small molecules that affect alternative splicing requires an effective, robust, and fast assay. Thus, high-throughput screening (HTS) is a promising and rapid methodology to identify potential modulators of the biological activity of the target from a large number of compounds.
In the present study, we describe the development and validation of a cell-based reporter HTS assay for the discovery of small-molecule inhibitors of PTB activity. This assay can also be used to screen a chemical library in search of activators of PTB. Small-molecule inhibitors of PTB may have use as research tools in the RNA splicing community. Importantly, identification of selective and potent inhibitors of PTB offers a promising strategy for molecularly targeted cancer treatment.
Materials and Methods
Cell Culture
The human ovarian cancer cell line A2780 was received as a generous gift from Dr. Thomas C. Hamilton (Fox Chase Cancer Center, Philadelphia, PA). Lenti-X 293T cells were purchased from Clontech (Mountain View, CA). All cell culture reagents were purchased from Mediatech (Manassas, VA). All cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum and 2 mM L-glutamine. Assay and optimization steps were performed in phenol red–free DMEM.
All compounds used in this study were dissolved into DMSO prior to their addition to cells. Compounds used for pilot screening were added 24 h after cell plating by pipetting 1 µL of either vehicle or small-molecule compound at a final DMSO concentration of 1% and incubated for 48 h in a humidified incubator at 37 °C with 5% CO2.
Microplates, Cell Seeding, and Plate Reader
Stock subcultures of parental A2780 and stable sublines were grown to ~70% to 80% confluency on standard cell culture-treated growth surfaces for 4 days and were then harvested using trypsin. Harvested cells were diluted in complete growth medium and counted. All cells were then seeded into microplates (CELLSTAR 96-well; Greiner Bio-One, Longwood, FL) at a density of 15 000 cells/well. All plates were seeded using the PerkinElmer Janus automated liquid handling system (PerkinElmer, Boston, MA). The seeded plates were incubated at room temperature for 45 min to allow the cells to settle uniformly on the growth surface of the well and then incubated overnight in a humidified incubator at 37 °C with 5% CO2 prior to beginning the assay readout. Following 48-h incubation with the compounds, the plates were analyzed with either the PerkinElmer EnVision (PerkinElmer) or BioTek Synergy 4 (BioTek Instruments, Winooski, VT).
Microarray Analysis
The splicing-sensitive microarray was purchased from Jivan Biologics (Greenbrae, CA); this specific product has been discontinued but updated, and similar products can be obtained from Jivan Biologics or ExonHit Therapeutics (Gaithersburg, MD). The array contains 116 205 unique probes representing 36 397 splice events (goo.gl/vGSRj). The array was used to discriminate between spliced, unspliced, and alternatively spliced RNAs of PTB-depleted and not-depleted samples. A2780 cells with and without PTB depletion were seeded in 100-mm dishes and incubated to 70% to 80% confluency. Total RNA was harvested in TRIzol reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. The probe labeling, array hybridization, washing, scanning, and data retrieval steps were performed by Jivan Biologics. The raw data obtained from Jivan Biologics were analyzed using SpliceFold software (goo.gl/TEPMX). The software generates a splice analysis report based on the preset filtering thresholds for signal probe intensity and splice-fold differences. The cutoff value for the significance of the signal probe intensity difference was set as p = 0.01. To identify differently expressed splice variants, we used the absolute splice score value cutoff of ≥0.3, which corresponds to a 2-fold difference for the ExpressionShort form/ExpressionLong form ratio. Results were confirmed by standard reverse transcriptase PCR (RT-PCR).
Construction of Minigene Reporter Plasmid
Human genomic DNA was extracted from A2780 cell lines and amplified by Platinum Pfx DNA Polymerase (Invitrogen) for 35 cycles with an annealing temperature of 58 °C and a 2-min extension time, using PTBP2 (NCBI Gene ID: 58155) specific primers E9F and E11R, with EcoRI and BamHI overhangs.
The resulting 2019-bp PCR product was selectively excised from a 1% agarose gel and purified using a Qiagen gel extraction kit (Qiagen, Valencia, CA). The resulting fragment was digested with the restriction enzymes EcoRI and BamHI (NEB, Beverly, MA) and cloned into pEGFP-N1 (Clontech), upstream of the reporter gene, to create pGreen. The DNA sequence of the ligation product was confirmed by sequencing on an ABI 3730XL DNA Analyzer (Applied Biosystems, Carlsbad, CA). The minigene containing the pGreen plasmid was transformed into DH5α-competent cells (Invitrogen). Plasmid DNA was prepared with NucleoBond Xtra plasmid DNA purification kit (Macherey-Nagel; Clontech), and the resulting plasmid was further confirmed by restriction digestion. The plasmid pEGFP-N1, encoding enhanced green fluorescent protein (EGFP) controlled by the cytomegalovirus (CMV) promoter (Clontech), was used as a source of the coding sequence of the pGreen minigene. The pGreen minigene was subsequently amplified using minigene-specific primers pGF and pGR, with MluI and SpeI overhangs, and the 2914-bp PCR product was subcloned into the pLV-tTR/KRAB lentiviral vector that results in LV-pGreen. The DNA sequence of the LV-pGreen plasmid was confirmed by restriction digestion and sequencing. The lentiviral vector was a generous gift of Dr. Didier Trono (University of Geneva, Switzerland).
Preparation of Lentiviruses Carrying Reporter Plasmid
The resultant lentiviral vector of LV-pGreen was packaged to generate viral particles. Lentivirus preparation and establishment of sublines of the ovarian cancer cells were done as described previously.14,21 LV-tTR harbors the EF-1α promoter within the 3′ LTR/SIN region and pGreen minigene as a reporter driven by this promoter. Lentiviruses were generated by cotransfection of Lenti-X 293T (Clontech) cells with three plasmids: a lentiviral vector plasmid plus pMD2.G (expressing envelop protein VSV-G) and psPAX2 (expressing packaging proteins). Media were changed 16 h after transfection, and the supernatants were harvested 48 h after transfection. Cell debris in the media was removed by 0.45-µm filtration following centrifugation at 1500 g for 10 min. The titers of lentiviruses in the media were determined by flow cytometry and ranged from 2 × 107 to 6 × 107 transducing units/mL. Packaging plasmids were also gifts from Dr. Didier Trono (University of Geneva, Switzerland).
Preparation of Lentiviruses Carrying Tetracycline-Inducible Expression Cassette of PTB shRNA
To manipulate PTB protein expression (positive controls in the assay), we used the tetracycline-inducible expression cassette of shRNA. The DNA fragments coding for PTB shRNA were generated by annealing two pairs of complementary oligonucleotides. The procedures for preparation of lentiviruses were detailed previously. 14
The Establishment of Stable Cell Lines
We established two new sublines using these lentiviral particles: A2780/pGreen and A2780/pGreen/Test. The former expresses a doxycycline-inducible PTB shRNA and pGreen reporter gene and was used as either a positive control (with doxycycline added) or a negative control (without doxycycline added); the latter, expressing the pGreen reporter alone, was used as a compound test cell line and/or a negative control. The establishment of stable cell lines expressing the reporter, pGreen alone, and pGreen and PTBshRNA was accomplished in multiple steps. To establish the A2780/pGreen/Test subline, parental cells (A2780) were transduced by lentiviruses carrying an expression cassette of the reporter minigene pGreen. Positive clones expressing pGreen were picked and enriched using flow sorting (Beckman Coulter MoFlo, Miami, FL).
To establish the A2780/pGreen subline, we first established cell lines transduced by lentiviruses LV-tTR/KRAB and then reinfected them with lentiviruses LV-TH/PTBshRNA. Clones expressing both KRAB protein and PTBshRNA were selected and expanded. The regulation by doxycycline of shRNA expression and KRAB protein expression in these clones was verified by measuring PTB expression by Western blotting. Later-picked clones were transduced by lentiviruses carrying an expression cassette of the reporter minigene pGreen. The isolated cell colonies were picked from the wells, transferred to 24-well plates, and grown in the presence or absence of 1 µg doxycycline/mL. Positive clones were identified by measuring both PTB expression by Western blotting and increased expression of green fluorescent protein in doxycycline-treated wells (data not shown).
Splicing Reporter Assay
We designed an assay to identify compounds that can modulate the splicing activity of the PTB protein. The assay uses fluorescence detection to monitor splicing of a green fluorescence protein–fused minigene, pGreen (derived from the PTBP2 genomic sequence, described above), to short or long splice variants (SV). Cellular PTB levels control the exclusion or inclusion of exon 10 in this minigene. Screening positives are compounds that block the splicing activity of PTB protein, therefore increasing the long SV. Induction of PTB shRNA by doxycycline completely blocks the splicing activity of PTB in this assay and is used as a positive control. DMSO alone is the best negative control for this assay during the HTS campaign. In detail, without any sequence modification, EGFP is in the reading frame when exons 9, 10, and 11 are spliced together (long form) in response to lower PTB activity, but it is out of the reading frame when exon 10 is skipped or repressed (short form) in response to higher PTB activity. Although the former translated into more EGFP, the latter expressed low levels of EGFP (
Fig. 1A
). We then developed a fluorescence cell-based assay that exploits this mechanism to allow HTS of small molecules for their ability to inhibit PTB activity. To perform the screening assay, experimental conditions were optimized to a 96-well plate format. To prepare the plates, A2780, A2780/pGreen(+)doxycycline, A2780/pGreen(–)doxycycline, and A2780/pGreen/Test cells were trypsinized, placed in each well of the 96-well plates, and allowed to attach overnight (1.5 × 104 cells per well [final]). The next day, DMSO was added to wells in plate columns 2 through 10 at a final concentration of 1% for HTS optimization; columns 1 and 12 contained positive controls (A2780/pGreen(+)doxycycline), negative controls (A2780/pGreen(–)doxycycline), and a background control (A2780). All wells had a final volume of 100 µL and 1% DMSO (nontoxic at this level). The plates were then placed back in the incubator for 48 h, after which they were removed and rinsed twice with phosphate-buffered saline. Fluorescence measurements were carried out on a PerkinElmer EnVision plate reader, using filters at 485/14 nm for excitation and 520/8 nm for emission. All liquid handling was carried out using an automated liquid handling system (PerkinElmer Janus). A more detailed HTS protocol is presented in

(
Data Analysis
We used positive (plus doxycycline) and negative (no doxycycline) signal controls in the plate wells to calculate Z′ values during the assay optimization to monitor assay robustness. Assays with a Z′ factor of 0.5 indicate that the assay is robust enough to identify inhibitors of PTB activity reliably. 22 Moreover, we used signal window (SW), coefficient of variance (CV), and assay variability ratio (AVR) values when distinguishing signals from positive control cell line activity from parental and negative control cell lines. 23
Results
PTB Regulates the Alternative Splicing of PTBP2 Pre-mRNA by Repressing Exon Recognition
Given the observation that PTB knockdown in A2780 causes suppression of tumor cell proliferation, suppression of anchorage-independent growth, and suppression of invasiveness, 14 we performed global exon array studies to identify potential regulatory targets responsible for these changes. The effect of PTB depletion on alternative pre-mRNA splicing and abundance was measured for 36 397 unique splice events in A2780 ovarian cancer cells using Jivan Biologics’ splicing-sensitive microarray. PTB depletion identified 317 genes whose splicing patterns were consistently altered more than 2-fold after PTB was depleted in two separate experiments ( Fig. 1B ). We validated differentially expressed splice variants identified by microarrays by conventional RT-PCR. Based on the confirmation data, high levels of PTB expression were associated with increased skipping of exon 10 in the PTBP2 gene (a PTB homolog, also known as brPTB or nPTB 24 ) in these A2780 epithelial ovarian cancer cells. By contrast, PTB depletion by shRNA knockdown introduces a premature stop codon (PTC), which causes nonsense-mediated decay (NMD) of the PTBP2 mRNA. This splicing event shown in Figure 1C and Figure 2A both creates the short- and long-form gene products of PTBP2.
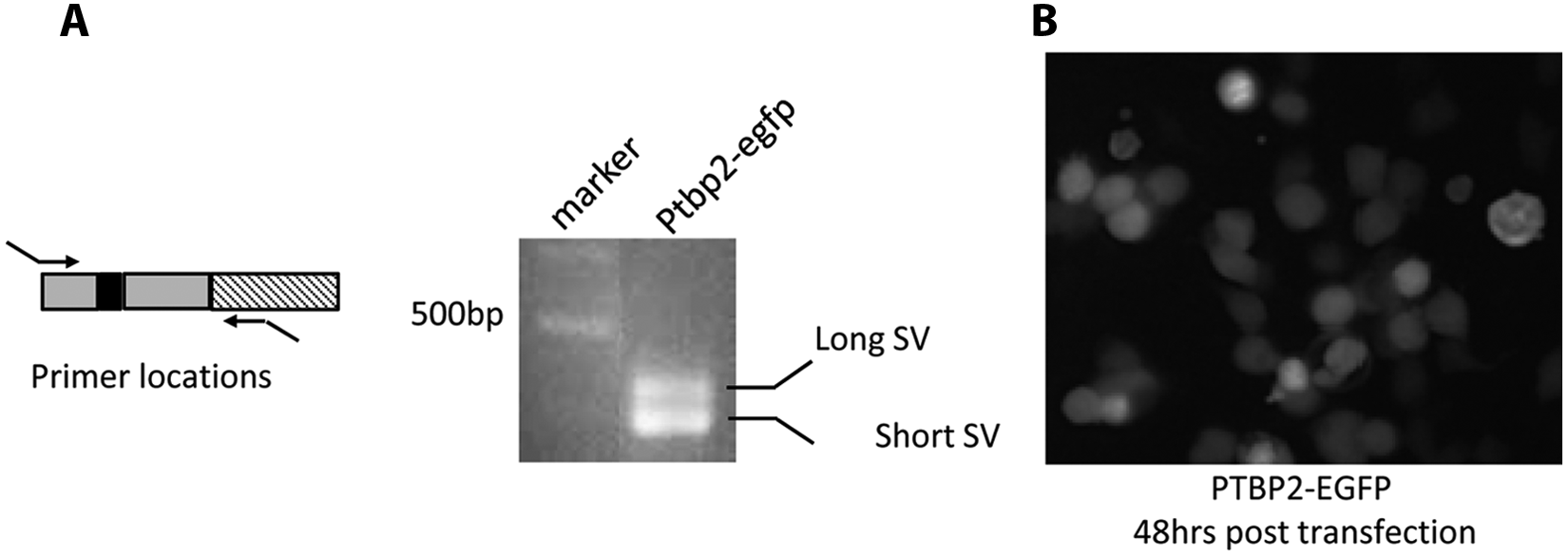
Polypyrimidine tract-binding protein (PTB)–responsive splicing fluorescence (FL) reporter system transfected into the cells to test its functionality. (
Cell-Based EGFP Reporter Assay Detects PTB Activity
Our previous findings13,14 support the idea that manipulation of AS by targeting PTB may have therapeutic potential for ovarian cancer treatment. To test this, we developed a cell-based fluorescent reporter assay to monitor PTB-mediated RNA splicing using an EGFP reporter as an indicator of PTB activity in live cells ( Figs. 1A , 2B ). This EGFP fused minigene reporter was designed based on differential splicing of a PTB target gene, identified from microarray analysis. This reporter also depends on the PTB levels in cells that were visualized by changes in reporter activity in terms of EGFP expression.
The microarray analysis and validation results ( Fig. 1B ) show that splicing of exon 10 of PTBP2 is regulated by PTB. Depletion of PTB by shRNA completely suppressed skipping of exon 10 (long form) and, importantly, led to an increase in PTB reporter activity. In contrast, high levels of PTB result in both the short and long forms being present in the transcript ( Fig. 3A ). We designed the reporter construct (called pGreen) so that the downstream EGFP will be in the reading frame when all three PTBP2 exons are spliced together (long form). When exon 10 is skipped, the disruption of the EGFP reading frame will lead to a decrease in PTB reporter activity (short form) by introduction of a PTC, which causes NMD of PTBP2 mRNA ( Fig. 3B ). Therefore, the higher reporter activity indicates the existence of the long variant with three exons included, and the lower reporter activity indicates the existence of the short variant with exon 10 skipped. When this construct is introduced into cells expressing high levels of PTB, such as Lenti-X 293T cells and A2780 cells, low EGFP levels are detected because both PTBP2 mRNA splice variants are generated in such cells. Figure 2B shows the experimental result of transfection of the construct into Lenti-X 293T cells. RT-PCR results indicated that the PTBP2 exons 9, 10, and 11 in the construct were coordinately spliced to form two distinct variants, the short and long forms, respectively ( Fig. 3B ), in response to altering the PTB levels. These results confirm the feasibility of using these constructs to detect the alternative splicing of PTBP2 exon 10 and PTB activity in our HTS system.

Functional minigene splicing analysis. (
Minigene Reporter Responses to Varying PTB Protein Levels
We performed several analyses to confirm that the regulatory elements in the minigene and EGFP expression were still functional in our engineered reporter cell lines. We first determined whether this reporter gene is responsive to different PTB levels. Following total RNA purification, we used vector-specific primers to amplify only the minigene reporter by RT-PCR (
Fig. 2A
). The results, in agreement with the microarray data, showed that the splicing pattern of the minigene reporter changes in PTB-depleted cells compared with PTB-expressing controls. We then purified the amplified PCR fragments from agarose gels and sequenced them, confirming that splicing of the minigene is still functional and that expression of EGFP can be detected (
Fig. 3B
). Next, we performed image-based analysis, using fluorescence (FL) microscopy (
Fig. 4A
). On the basis of the image analyses, we observed more than a 2-fold increase in PTB reporter activity in PTB-depleted cells compared with controls (
Fig. 4B
), and this was due to alternative splicing of the minigene. In addition to image-based analysis, we also performed flow cytometry experiments to validate our observations. Flow cytometry analysis resulted in similar changes in the reporter activity pattern, as observed in image-based analysis (

Quantitation of fluorescence (FL) intensity change in engineered cells after polypyrimidine tract-binding protein (PTB) knockdown (KD). (
Establishing a Reliable High-Throughput Assay in 96-Well Plates
Assay and plate parameters
To establish maximum signal strength and minimize intraplate and interplate variability, we prepared several plates containing 1% DMSO, as described in Materials and Methods. This validation was completed using all three engineered cell lines (A2780/pGreen(+)doxycycline), A2780/pGreen(–)doxycycline, and A2780). The assay plates showed a coefficient of variation (CV) of 6.2% for the inducible positive controls and 4.5% for the negative controls. We next addressed the assay performance measures (APMs) for our cell-based assay in terms of signal window (SW), Z′ factor, and AVR values to assess whether our assay is suitable for HTS.
23
We conducted multiple test runs to examine the SW, AVR, and Z′ of our HTS assay. We used a fixed number of controls to mimic a real run during the evaluation of the assay (six samples of positive and negative controls per plate). We determined the Z′ factor,
22
which measures the dynamic window between total signal and background, taking into account the errors associated with each, as 0.63 ± 0.09 (n = 9). Overall, we assessed nine different reading conditions during these test runs and calculated means and standard deviation of positive and negative controls, SW, AVR, and Z′, as shown in
Controlling for DMSO toxicity
The common way of preparing stock libraries of compounds in 100% DMSO solution raises toxicity concerns in a cell-based assay. Using the proper DMSO controls in the chemical screen minimizes the possible DMSO effect on the screens of chemicals. The degree of chemical cytotoxicity depends on the nature of the cell background, the concentration of the chemical, and the length of exposure.
25
We assessed the effects of DMSO on assay cell growth and reporter activity in two experiments to optimize the final chemical concentration to use in our HTS assay. To optimize the final DMSO concentration, medium with DMSO concentrations ranging from 0% to 1.5% was applied to the test cell line (A2780/pGreenNI) and control cell line (A2780/pGreen) for 48 h. We found that cell viability and reporter activity were not affected significantly with increasing DMSO concentrations (
Kinetics of PTB knockdown
The kinetics of PTBP2 pre-mRNA processing (splicing) in cancer cells is not known, and this prevented us from estimating the exposure time to the compounds needed in our HTS assay. To better understand the kinetics of PTBP2 mRNA splicing, we used a simple time course knockdown approach. We employed PTB-targeting synthetic oligonucleotides. After transfecting the small interfering RNA (siRNA) oligos into the A2780 cells, RNA samples were collected at 0, 2, 5, 9, 18, and 48 h to analyze splicing products of the PTBP2 reporter minigene and also mRNA levels of PTB ( Fig. 5A ). RNA samples were reverse transcribed into complementary DNA (cDNA), and these cDNAs were used as templates in the PCR reaction mix. Resultant PCR products were separated by agarose gel electrophoresis and quantified ( Fig. 5B ). The PTB siRNA time course study of the minigene splicing activity showed an increase in the long-form expression of PTBP2, whereas PTB mRNA was downregulated ( Fig. 5A ). On the basis of this time course study, we selected an incubation time of 48 h for further experiments that produced sufficient signal for fluorescence readout.

Time course study of polypyrimidine tract-binding protein (PTB) knockdown effect on PTBP2 messenger RNA (mRNA) long-splice form formation. (
Assay performance
During the assay performance evaluation phase, the calculated SW, AVR, and Z′ values met our quality control criteria, as seen in
Discussion
Our previous results13,14 that knockdown of PTB expression causes suppression of tumor cell proliferation, anchorage-independent growth, and invasiveness all strongly support the notion that PTB is important in maintaining ovarian tumor cell growth 14 and is associated with the degree of tumor malignancy. 21 At present, it is not clear what mechanisms mediate these effects. However, current knowledge about the targets of PTB cannot entirely explain these observations. Despite these gaps in our knowledge, our results clearly support the idea that PTB has potential as a therapeutic target for the treatment of ovarian cancer and possibly other cancers in which it is overexpressed.
Using a minigene reporter approach, we have established herein a rapid, reliable, and reproducible cell-based HTS assay system that can be used to identify bioactive small molecules capable of modulating PTB activity. To the best of our knowledge, ours is the first report that outlines the development of an HTS campaign that uses an engineered minigene to monitor PTB-mediated splicing events in ovarian cancer cells. We first established cell lines stably expressing the reporter minigene and appropriate control vectors to identify modulators of PTB activity, and we then verified and optimized these cell lines to use in our HTS campaign. This approach represents an efficient system for the detection of novel compounds, applicable for both academic and industrial purposes. To successfully develop and execute an efficient, rapid, and reproducible cell-based HTS assay in the field of antitumor drug discovery, one must have access to (1) an automated screening technology platform and (2) an accurate and reliable reporter system, which, in this case, was an A2780 stable cell line that expresses the minigene reporter. Traditional techniques using 12- or 24-well plates have been shown to be more problematic in screening big chemical libraries than are the more high-density plate formats (≥96 wells). In addition, small assay volumes are required for maximum efficiency to minimize the cost of the assay, assay time, and reagent consumption. Recent advances in HTS technologies have made it possible to execute cell-based HTS campaigns with smaller formats. Use of automation systems for cell plating, compound transfer, and plate reading further maximizes efficiency.
A cell line carrying an accurate and reliable reporter is needed to screen large libraries of small molecules efficiently. In our case, the selection and optimization of the most appropriate minigene splicing reporter cell line are very important. To successfully perform HTS using 96-well plates, several characteristics of the host cell line must be taken into account, such as basal PTB levels, doubling time, transduction efficiency for readout of the reporter gene, growth characteristics in the microwell environment, and the kinetics of splicing. We chose to use the A2780 cell line because of its high level of PTB expression and high transduction efficiency. 14 The level of PTB expression appears to be very high in this cell line, although the precise comparison of expression levels between various cell lines is difficult because of differing conditions in different laboratories (e.g., the source of the cell lines, antibodies). We further chose a reporter bearing the EGFP gene because it can be measured easily, requires no additional reagents, and has a suitably long half-life. 26 Indeed, we obtained a high signal-to-background ratio (~1:20) in the 96-well plate format, indicating the suitability of this system for HTS assays. 27
To facilitate the detection of splicing repressors, several assays have been developed,28–31 including a cell-based assay 31 with a luciferase reporter gene. However, this replicon model has several disadvantages: (1) It targeted the spliceosomal subunit that consists of multiple associated proteins, making it difficult to predict the real target; (2) it required additional processing to measure the reporter itself; and (3) because the cell lines were transiently transfected, it required extensive optimization prior to screening, making it difficult to control the amount of the gene transfected. Accordingly, stable cell lines bearing a minigene fused to an EGFP reporter gene, such as we have described herein, appear to be better suited for use in our HTS campaign to target a specific splicing factor.
An important consideration for our HTS campaign is that many small molecules can be cytotoxic to cells, and this cytotoxicity can be mistaken for inhibitor activity by decreasing the reporter signal when it only decreases cell viability without a direct effect on reporter activity, thus leading to false-positive results. We therefore decided to incorporate an additional step in our assay to measure cytotoxicity in parallel with our primary HTS splicing readout. Because of the nature of the original reporter assay we developed, we did not need to design a new assay; rather, we multiplexed our assay format by measuring basal EGFP expression from the A2780/pGreen/Test cell lines, which was 5 to 10 times weaker than the induced EGFP levels in the positive control cell lines. The weak EGFP signal served as a viability marker for the cells treated with the compounds in our HTS. This additional analysis was performed to minimize identifying erroneous, false-positive hits from increased or decreased fluorescence signals due to increased or decreased cell viability, respectively.
Despite the substantial potential of fluorescent protein–based reporter applications to measure biological interactions in a cell-based, high-throughput format, there are also difficulties encountered when using fluorescent proteins. For example, fluorescent compounds can interfere with fluorescent protein assays. 32 Because this common fluorescence interference can be a problem when using diverse compound libraries that may contain members that fluoresce in the excitation and emission spectra of EGFP, we further modified the detection format of our HTS assay to measure the progress of the splicing reaction kinetically, as opposed to collecting a single end-point read. Although multiple measurements can increase the overall assay time, a fast-scanning reader, such as we have, in combination with an automated system can allow rapid and repeated measurements of multiwell plates without significantly slowing the overall plate processing speed. Importantly, the collection of a two-point time course allows the effects of fluorescent but otherwise inert library members to be eliminated to reveal the true reaction course. 32 Because the first time point values (when the compounds are added prior to initialization of splicing) associated with each compound well are stored in the output data, a further analysis can be performed to flag interfering fluorescent library members. In our HTS study, we collected two points per well to reduce further assay interference, because when we performed our analyses as a single end-point read only, our hit rates were unacceptably high, primarily due to false-positive results from the fluorescent compounds.
A cell-based HTS method alone is itself not sufficient to complete a screening campaign. In a proper design, hits are evaluated in secondary assays that test the ability of the hit to modulate a particular biological event that is distinct from the primary screening assay. Therefore, an appropriate secondary assay must be incorporated to the campaign to verify hit compounds identified from primary screening. To evaluate these compounds, our secondary screen will use our previously defined gene signature ( Fig. 1C ) that was generated by using a splicing-sensitive microarray and is unique to PTB-depleted cells. As part of the screening process, RT-PCR will be used to detect the effects of the compounds on other validated PTB targets.
Like other traditional cell-based HTS methods, our screening campaign was designed to be executed at a single fixed concentration per compound. The generation of the dose-response curves for each of the compounds screened in a primary screen would be inordinately burdensome even for the most advanced screening centers. Thus, it is considerably more practical and effective to generate the dose-response data only for those compounds identified as hits in the primary screen and validated in the secondary validation screens. Moreover, orthogonal assays will be used to further validate the selected hits. These involve examination of the hits on (1) PTB levels by Western blot, (2) ovarian cancer cell growth, and (3) multiple PTB-regulated alternative splicing events, using a splicing-sensitive microarray as we have shown in Figure 1B .
Although our cell-based HTS system represents an effective screening method to identify potential modulators of PTB splicing activity, there is a potential limitation to the use of this reporter as a surrogate marker of PTB activity in all cell lines because of their variable levels of PTB expression. By using cell lines expressing high levels of PTB in our study, we have addressed this particular limitation.
In conclusion, we have presented herein a PTB-specific cell-based fluorescence splicing assay that can quantitate inhibition of PTB activity by using one of its target genes, PTBP2, that we identified previously. We also incorporated a built-in cell viability control that exploits the steady and low level of EGFP expression at the basal state as an indicator of viability. Finally and importantly, our assay reported herein provides a unique way to measure the inhibited target activity. Thus, the repression of PTB activity results in the increase of reporter readout so the assay allows one to easily discern nontoxic and PTB-specific inhibitory substances.
In sum, we have developed reliable HTS methods that will accelerate the discovery of small molecules that modulate PTB splicing activity. We are currently optimizing the assay to extend the screening to larger libraries that are available at the Molecular Libraries Screening Centers Network. Moreover, because this protein has been linked to ovarian 14 and breast 33 tumors, as well as glioblastomas, 15 this platform has potential to generate new therapeutic leads to treat these diseases.
Footnotes
Acknowledgements
We thank Dr. Didier Trono (University of Geneva, Switzerland) for his generous gift of lentiviral vectors LV-THM and LV-tTR/KRAB-Red as well as plasmids pMD2.g, pMDLg/pRRE, and pRSV Rev. We are most grateful to our colleague, Martina Vaskova, for her outstanding administrative, technical, and intellectual assistance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Cancer Institute grant R01 CA138762 to WTB. It was conducted in a facility constructed with support from the NCRR NIH grant C06RR15482.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.