
Abstract
Tumor cell subpopulations called cancer stem cells (CSCs) or tumor-initiating cells (TICs) have self-renewal potential and are thought to drive metastasis and tumor formation. Data suggest that these cells are resistant to current chemotherapy and radiation therapy treatments, leading to cancer recurrence. Therefore, finding new drugs and/or drug combinations that cause death of both the differentiated tumor cells as well as CSC populations is a critical unmet medical need. Here, we describe how cancer-derived CSCs are generated from cancer cell lines using stem cell growth media and nonadherent conditions in quantities that enable high-throughput screening (HTS). A cell growth assay in a 1536-well microplate format was developed with these CSCs and used to screen a focused collection of oncology drugs and clinical candidates to find compounds that are cytotoxic against these highly aggressive cells. A hit selection process that included potency and efficacy measurements during the primary screen allowed us to efficiently identify compounds with potent cytotoxic effects against spheroid-derived CSCs. Overall, this research demonstrates one of the first miniaturized HTS assays using CSCs. The procedures described here should enable further testing of the effect of compounds on CSCs and help determine which pathways need to be targeted to kill them.
Introduction
The American Cancer Society estimated that more than 240 000 new cases of male prostate cancer were diagnosed in 2011, and 30 000 men will die due to this disease. Localized prostate cancer responds well to resection, chemotherapy, and radiation, but overall survival rates are low as a result of colonizing metastases. A lower number of Americans were diagnosed with pancreatic cancer in 2011 in the United States with 44 000 new patients, 1 yet the disease remains the fourth leading cause of cancer death overall. In the same year, 37 000 people were predicted to die as a result of this disease, 1 which is 85% of all diagnosed patients. Since 1975, the 5-year survival rate for pancreatic cancer has improved only from 3% to 6%, and in fact, pancreatic cancer is the only one of the top 10 cancer killers that still has a 5-year survival rate in the single digits. Surgical resection is the best method for long-term survival in patients, but only about 15% of the patients are diagnosed early enough to be eligible for surgery, and furthermore, 80% of those patients who will undergo surgery will have a recurrence of the disease within 2 years. 1 This percentage remains high due to the aggressive nature of pancreatic cancer and the ability of highly invasive cells to resist current treatment regimes.2–4
In both pancreatic and prostate cancers, highly invasive cells are able to survive and metastasize to other vital organs, which is uniformly fatal in patients. 2 A vast majority of the patients diagnosed with pancreatic cancer receive the nucleoside analogue drug gemcitabine as part of their treatment regime, and those with prostate cancer receive the cell cycle inhibitor docetaxel.5,6 Recent evidence suggests that a small population of cells, referred to as tumor-initiating cells (TICs) or circulating tumor cells (CTCs), within a heterogeneous tumor have an enhanced capacity to form a tumor; are responsible for propagation, relapse, and metastatic dissemination; and are the root cause of patient resistance to treatment and ultimate death. However, because these tumor-initiating cells also possess certain stem cell–like properties (e.g., quiescence, self-renewal, asymmetric division, and multidrug resistance), permitting them to drive tumor growth and evade conventional therapies, they have also been called cancer stem cells (CSCs). To date, CSCs have been isolated from almost every solid tumor type.7–11 In the past few years, CSCs have been isolated from cancers by fluorescence-activated cell sorting (FACS) selecting for expression of CD44, CD24, ESA, or CD133. 12 In addition, CSCs can be isolated by generating spheroids using specialized culture conditions and highly defined media called stem cell media (SCM).13–16 The spheres express higher levels of the stem cell markers indicated above and demonstrate higher tumorigenic potential in animals compared with total cells.15,16
Because of the limited treatment options for aggressive types of many cancers, we sought to find compounds capable of killing the CSC populations in spheroid-derived model systems of the prostate and the pancreas. 17 Using a 1536-well microplate format and a cell growth assay, an in-house small-molecule collection consisting of compounds targeting oncology-relevant pathways/mechanisms of survival was screened. The screen was implemented by testing the compounds in dose-response format, which allowed for a hit selection process that included both potency and efficacy to efficiently identify compounds with potent cytotoxic effects for CSCs. A few compounds were able to inhibit the growth of the CSCs and the parental adherent cells from both cancer types with high potency and efficacy. The initial results were further validated in a functional assay of CSC invasion using Matrigel-coated Boyden chambers. Finally, we used a highly aggressive mouse breast cancer cell line, 4T1.2, which has been stably infected with a Nanog-GFP reporter as a model system to determine if compounds affected the “stemness” of the cells by measuring effects directly on a stem cell reporter. This research demonstrates one of the first quantitative high-throughput screening (qHTS)–based assays using nonengineered CSCs. Overall, the determination of which pathways need to be inhibited to produce toxicity against CSCs will result in the development of novel therapies for aggressive forms of cancer and will specifically result in targeting metastasis and recurrence.
Materials and Methods
Cell lines and reagents
PANC1 human pancreatic cancer and LNCaP human prostate cancer cell lines were obtained from ATCC (Manassas, VA) and cultured according to the manufacturer’s instructions (Cell Line Verification Test Recommendations, ATCC Technical Bulletin No. 8 [2008]). The mouse breast cancer line 4T1.2 Nanog-GFP was provided by Dr. Thomas Sayers from NCI-Frederick (Frederick, MD) and were grown in RPMI + 10% fetal bovine serum (FBS) (see Supplemental Methods for its construction). SCM was prepared as previously described. 18 Salinomycin was purchased from Sigma (St. Louis, MO), and all other chemicals (bortezomib, CMP1, and CMP1-S) were purchased from Selleck (Houston, TX).
Sphere formation assays
LNCaP and PANC1 spheres were generated as previously described.18,19 Additional information is available in the Supplemental Methods section.
Proliferation assays
Assays were conducted in sterile, tissue culture–treated 1536-well white solid-bottom plates (catalog number 789173-F; Greiner Bio-One, Monroe, NC). A total of 200 cells per well in 5 µL of SCM + KO + ITS were seeded using a Multidrop Combi Reagent dispenser with a small pin cassette (Thermo Scientific, Fisher Scientific, Fair Lawn, NJ). Immediately after dispensing the cells, 23 nL compound solution in DMSO was transferred using a Kalypsys (San Diego, CA) pintool. The plates were then covered with stainless steel Kalypsys lids and placed into an incubator at 37 °C, with 5% CO2 and 95% relative humidity. The plates were incubated for 48 h, and then 3 µL CellTiter-Glo reagent assay (Promega, Madison, WI) was added using a BioRAPTR (Beckman Coulter, Brea, CA). Plates were incubated for 30 min at room temperature and spun at 1000 rpm, and relative luminescence units (RLU) were quantified using a ViewLux (PerkinElmer, Waltham, MA).
Mechanism Interrogation PlatE (MIPE) compound library
The library used in these studies is an internal collection of 112 high-value small molecules known to modulate oncology targets, pathways, and phenotypes, referred to as the MIPE-oncology library (MIPE: Mechanism Interrogation PlatE) (see Supplemental Methods section for additional information, and a full list of compounds is available upon request).
Quantitative high-throughput screen (qHTS)
For the screen, the MIPE library compounds were transferred to columns 5 to 48, and controls were added in columns 1 to 4 of the assay plate. Columns 1 and 2 contained plain DMSO, whereas columns 3 and 4 contained 2-mM solutions of the protease inhibitor bortezomib or the antibiotic salinomycin in DMSO (final concentration 9 µM). Compounds were tested as dose responses starting at a stock concentration of 10 mM (final concentration 46 µM) in DMSO and diluted 3-fold, also with DMSO. The library was tested at 12 compound concentrations for qHTS as described previously. 20 Relative luminescence units for each well were normalized to the median RLUs from the DMSO control wells as 100% viability and median RLUs from the salinomycin or bortezomib control wells as 0% viability. Additional information is available in the Supplemental Methods section.
Hit selection from qHTS
Hits were selected based on two methods: (1) a single % viability parameter at the compound dose that produced maximum cell killing. A compound that produced ≥70% cell killing (≤30% cell viability) at the dose that produced maximum killing was considered a hit. (2) Curve response class (CRC) classification from dose-response HTS, in which normalized data were fitted to four-parameter dose-response curves using a custom grid-based algorithm to generate a CRC score for each compound dose response.20,21 CRC values of −1.1, −1.2, −2.1, and −2.2 are considered high-quality hits; CRC values of −1.3, −1.4, −2.3, −2.4, and −3 are inconclusive hits; and a CRC value of 4 is for inactive compounds. See Supplemental Methods for additional information.
Matrigel invasion assay
Matrigel-coated 24-well inserts (8 µm pore size) were purchased from BD Biosciences Clontech (Palo Alto, CA), and the assay was done as described previously. 17 A detailed assay protocol is available in the Supplemental Methods section.
High-content imaging Nanog-GFP assay
A 1536-well high-content assay to measure Nanog-GFP levels in 4T1.2 cells was developed using an ArrayScan VTI (Thermo Scientific). A detailed assay protocol is available in the Supplemental Methods section.
Results
Development of a 1536-Well Microplate CSC Proliferation Assay Using CellTiter-Glo
A major challenge to using cancer stem cells for drug discovery is their production in quantities that enable their testing for screening of compound libraries. Limited number of CSCs can be isolated from bulk cancer cell populations by sorting for specific cell surface stem cell antigens, including CD44, CD24, and CD133.7,8,22 Alternatively, protocols have also been developed for the isolation of CSCs from cell lines forming spheroids by using stem cell–specific growth media and nonadherent surfaces. The spheroid technique affords for the production of large amounts of CSCs and therefore enables the availability of these cells for the use in HTS. In this work, we adapted previously described methods to obtain pancreatic and prostate CSC spheroids from cancer cell lines (
Screening the MIPE of Oncology Pathway Compounds
The spheroid CSC 1536-well plate growth assay was used to screen the National Center for Advancing Translational Sciences (NCATS) MIPE library of small molecules, which consisted of 112 unique compounds targeting oncology-relevant pathways/mechanisms of survival. The compounds were tested at 12 doses, starting at 46 µM and diluted 3-fold, for their effect on growth of both PANC1 and LNCaP cells, both grown as spheres (CSCs) and adherent cells (original parental cells from which CSCs were obtained). Results were analyzed using two methods: % viability (corresponding to the % viability at the compound dose that produced maximum killing) and curve fit to the compound dose responses to generate CRC scores (see Materials and Methods). Analysis of the results comparing the % viability parameters for the different cell types and growth conditions showed robust correlation (R2 = 0.47) between the effects of compounds on cells grown as spheres versus adherent cells (
Fig. 1A
). In contrast, we observed a lack of correlation (R2 = −0.81) between the activity of the compounds on LNCaP versus PANC1 cells (
Fig. 1B
), suggesting that the cancer type, rather than growth mode, drove the differential cytotoxic effect of the compounds in the collection. A hierarchical clustering of the % viability values for the four different parameters is shown in
Figure 1C
. It again shows that clustering of compounds is driven mostly by the activity in cancer cell type, and most of the compounds are selective for their effect on either LNCaP or PANC1, regardless of whether the cells are grown as spheres or adherent cells. A small number of compounds are pan-active (meaning they were effective on both adherent cells and spheres and in both lines LNCaP and PANC1), and very few appear to be selective for just the spheres (
Fig. 1C
). A stringent cutoff of ≤30% viability was chosen to select compounds as hits. This activity cutoff for hit selection was based on published data highlighting the resistance of CSCs to conventional therapies,24,25 as well as based on the statistical parameters of the assay, especially for the sphere assay, where the mean−3*standard deviation of the sample field for a DMSO plate was close to 30% viability (
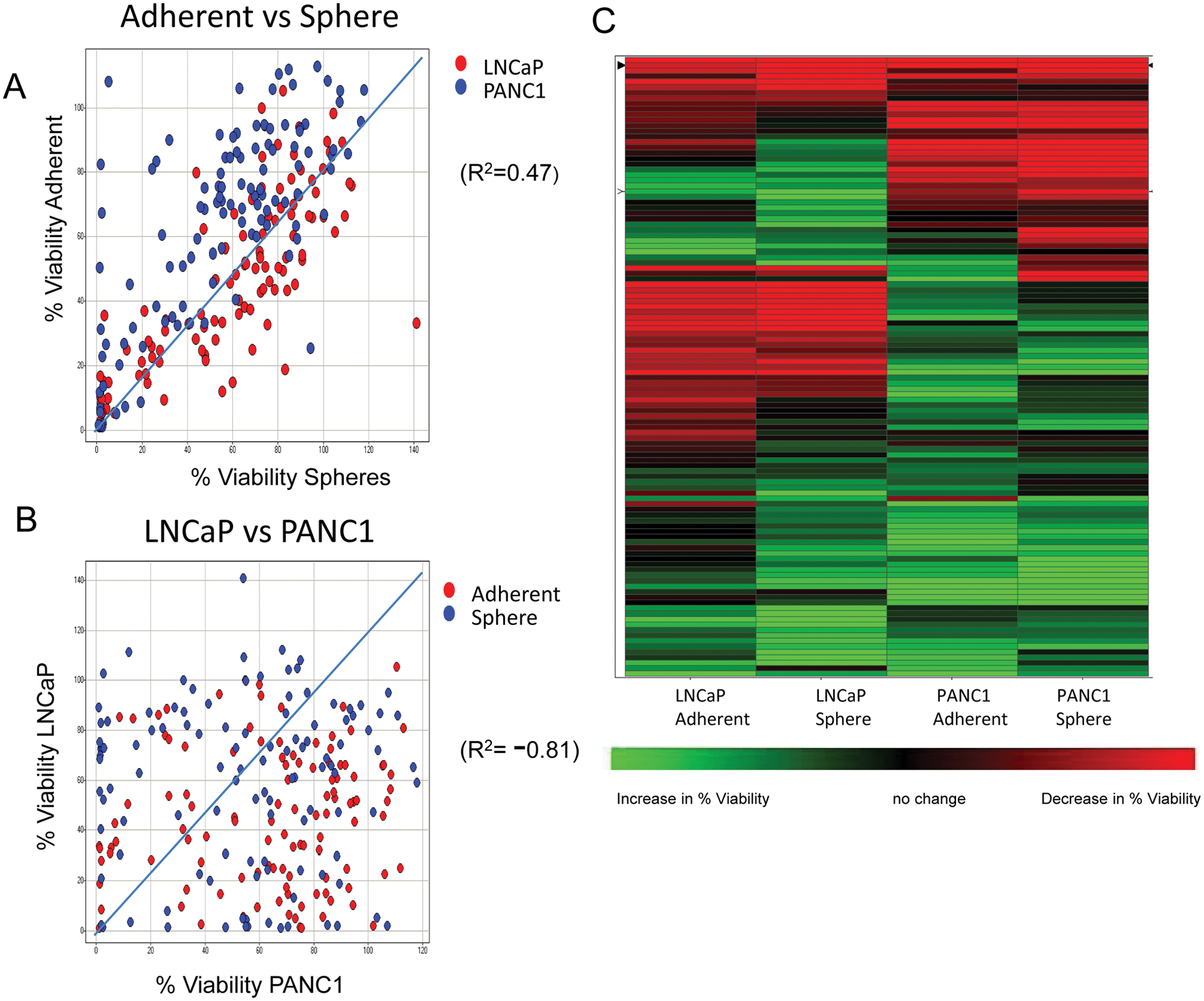
Results from quantitative high-throughput screening (qHTS) analyzed using % viability. (
A drawback of the traditional hit selection method based on a single % viability parameter to measure compound activity is that it selects compounds based mostly on efficacy and does not efficiently discern compounds based on potency. In this case, because the compounds were tested in dose responses, we were able to score each compound in dose response to a CRC,
20
which is a measure that includes potency, efficacy, and reliability of the data and estimates an IC50 value directly from the primary screen. Compounds that might look equally potent by % viability might in fact have a degree of selectivity that is only evident when comparing their CRCs and IC50 values. The distributions of CRCs from the four parameters are shown in
Figure 2
. For the purposes of this study, we only considered active compounds those with high-quality curve classes (−1.1, −1.2, −2.1, and −2.2). In general, the CRC distribution was very similar for both cell lines and growth conditions, suggesting that neither of the assay conditions tested was much more susceptible to compound treatment than the others. Any overlaps are visualized in Venn diagrams (
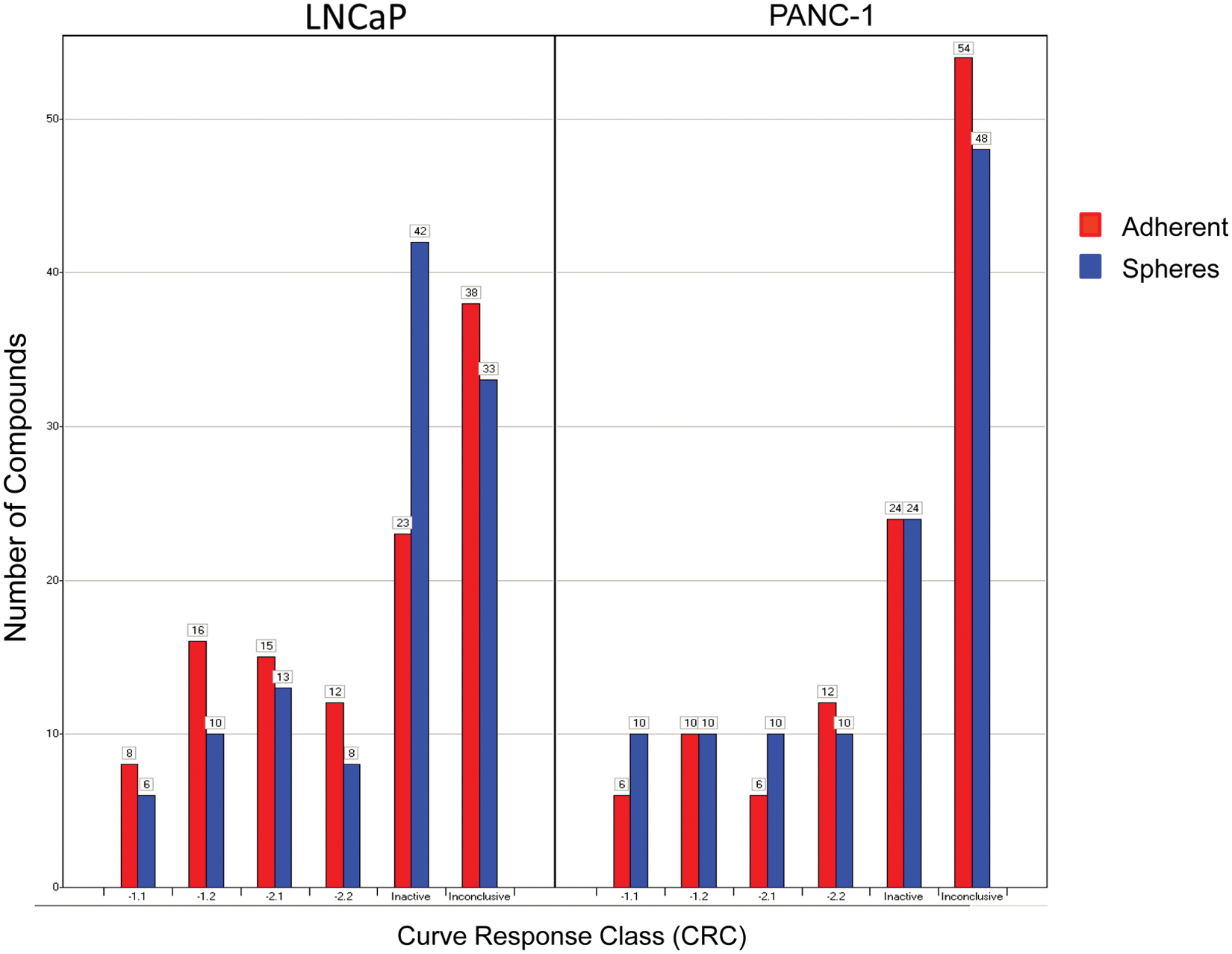
Results from quantitative high-throughput screening analyzed using curve response class (CRC) fitting, showing the CRC distribution of the number of compounds from both the LNCaP and PANC1 cell lines for both adherent (red) and sphere (blue) populations. CRCs −1.1, −1.2, −2.1, and −2.2 are considered active, and CRC 4 is inactive. All other groups are clustered as inconclusive. A more detailed description of the CRCs can be found in the Supplemental Methods section and referenced in Inglese et al. 20 and Wang et al. 21
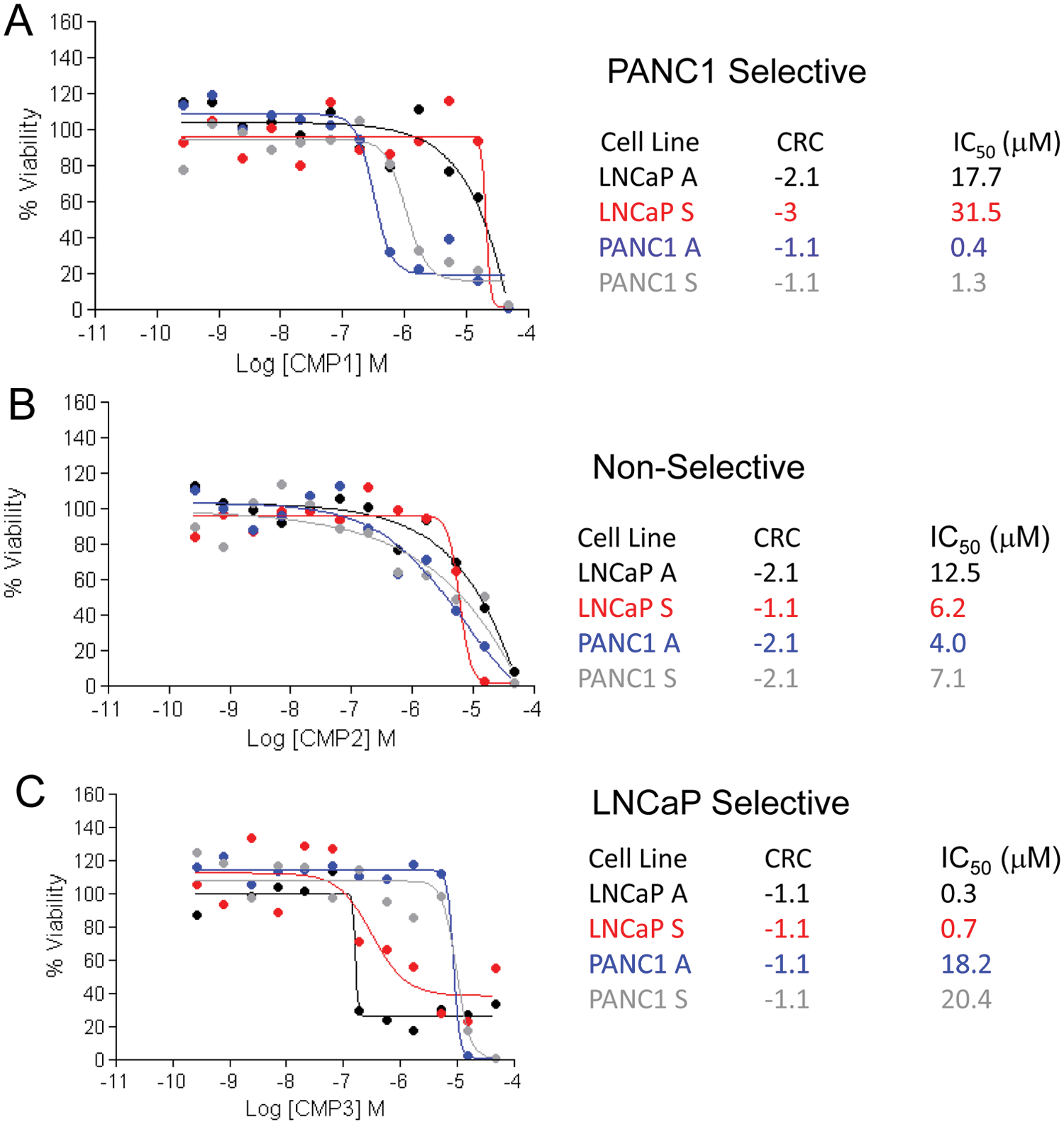
Dose-response curves of (
As discussed in the introduction, gemcitabine is the current standard of care treatment for pancreatic cancer. Gemcitabine is included in the MIPE collection and was therefore of interest to investigate the effect of this drug in the screen performed (
A Secondary Functional CSC Matrigel Invasion Assay to Test Compounds Identified in the Primary Proliferation Assay
To determine whether the compounds selected from the proliferation assay could also inhibit the invasive ability of CSCs, a Matrigel invasion assay was used as a secondary assay ( Fig. 4 ). In this assay, cells migrate toward the SCM media, and after 24 h under cell culture conditions, the noninvasive cells are removed and the invasive cells are fixed, stained, and counted.24,25 The noninvasive cells are removed prior to staining to determine viability and to demonstrate negligence permeability of the compounds across the matrix (data not shown). Compared with DMSO ( Fig. 4B ), bortezomib ( Fig. 4C ) and salinomycin ( Fig. 4C ) were able to significantly inhibit invasion at both the lower and higher concentrations tested ( Fig. 4A ). At the higher concentration, CMP1 ( Fig. 4C ) and CMP1-S, a compound inhibiting the same target ( Fig. 4C ), were both able to inhibit invasion toward the SCM ( Fig. 4A ), demonstrating that the proliferation-based assay used for primary screening had the ability to select for compounds capable of inhibiting the invasive ability of CSCs.
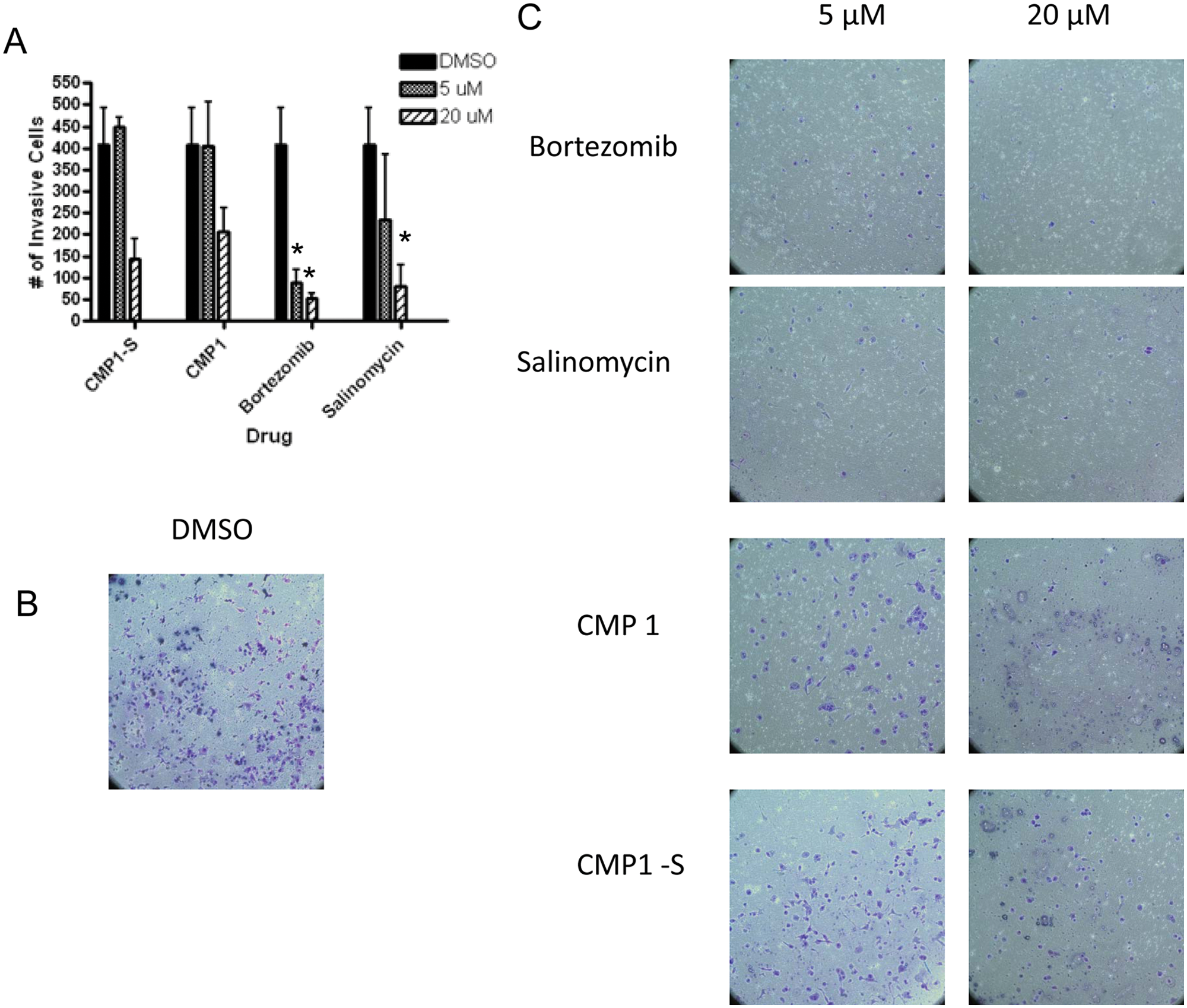
Results from the PANC1 Matrigel cell invasion assay. PANC1 cells were allowed to invade Matrigel membrane toward stem cell media (SCM) for 24 h and then fixed and stained. Two independent experiments were performed and averaged to generate error bars. *Indicates statistical significance compared with DMSO using a pairwise Student t test (p < 0.05). (
A High-Content–Based NANOG-GFP Assay for Measuring “Stemness” of CSCs
A possible mechanism by which compounds could reduce resistant CSCs to treatment is by modulating their stem cell–like characteristics. To determine the ability of the compounds to regulate the stem cell–like properties of CSCs, we used a high-content imaging assay using a 4T1.2 mouse breast cancer cells with a Nanog-GFP reporter. In the growth conditions used, about 30% to 40% of the 4T1.2 Nanog-GFP cells are GFP+, demonstrating they express Nanog (Thomas Sayers Laboratory–NCI Frederick, unpublished data). If the GFP+ cells are then sorted and placed back in culture, the same percentage of cells will remain GFP+ and are much more metastatic when seeded in an animal (Thomas Sayers Laboratory–NCI Frederick, unpublished data). The selection of Nanog as a marker for CSCs was based on literature supporting the role of this gene in maintaining the CSCs phenotype.26–30 To analyze the GFP content in the 4T1.2 cells after compound treatment, we fixed cells and stained the nuclei with Hoechst dye to determine cell number. A nuclear-masked–based compartmentalization algorithm was used to measure nuclear GFP signal from each cell. A mean average nuclear GFP signal per cell for each compound treatment was computed by averaging the nuclear GFP signal from each cell counted in a well. Data were normalized to DMSO-treated cells. 67 compounds reduced cell number to ≤30% (similar cutoff used for the cell proliferation assay), including the two pan-active hits identified using the % viability method and the five pan-active hits found from the CRC method (data not shown). When analyzing the data from the Nanog channel by looking at average GFP expression per cell, very few compounds effectively reduced the mean nuclear Nanog-GFP signal per cell. Figure 5A shows a dose-response profile from a compound (CMP4) that is able to reduce both the Nanog-dependent GFP signal per cell and the signal in the Hoechst channel. The images for three different concentrations of CMP4 treatment in both channels are shown in Figure 5B . CMP1, a compound selected as a pan-active hit from the % viability analysis, shows a decrease in cell viability with an increase in mean nuclear GFP signal per cell before no signal is detected at high compound doses because no cells survive the treatment. When further analyzing the images, it can be observed that the majority of the cells are dying with compound treatment ( Fig. 5C , D ), but the few surviving cells are high expressers of Nanog ( Fig. 5C , D ). We currently do not have an explanation for this effect, and it is under investigation that these surviving cells could be drug-resistant cells that still remain alive albeit with treatment.
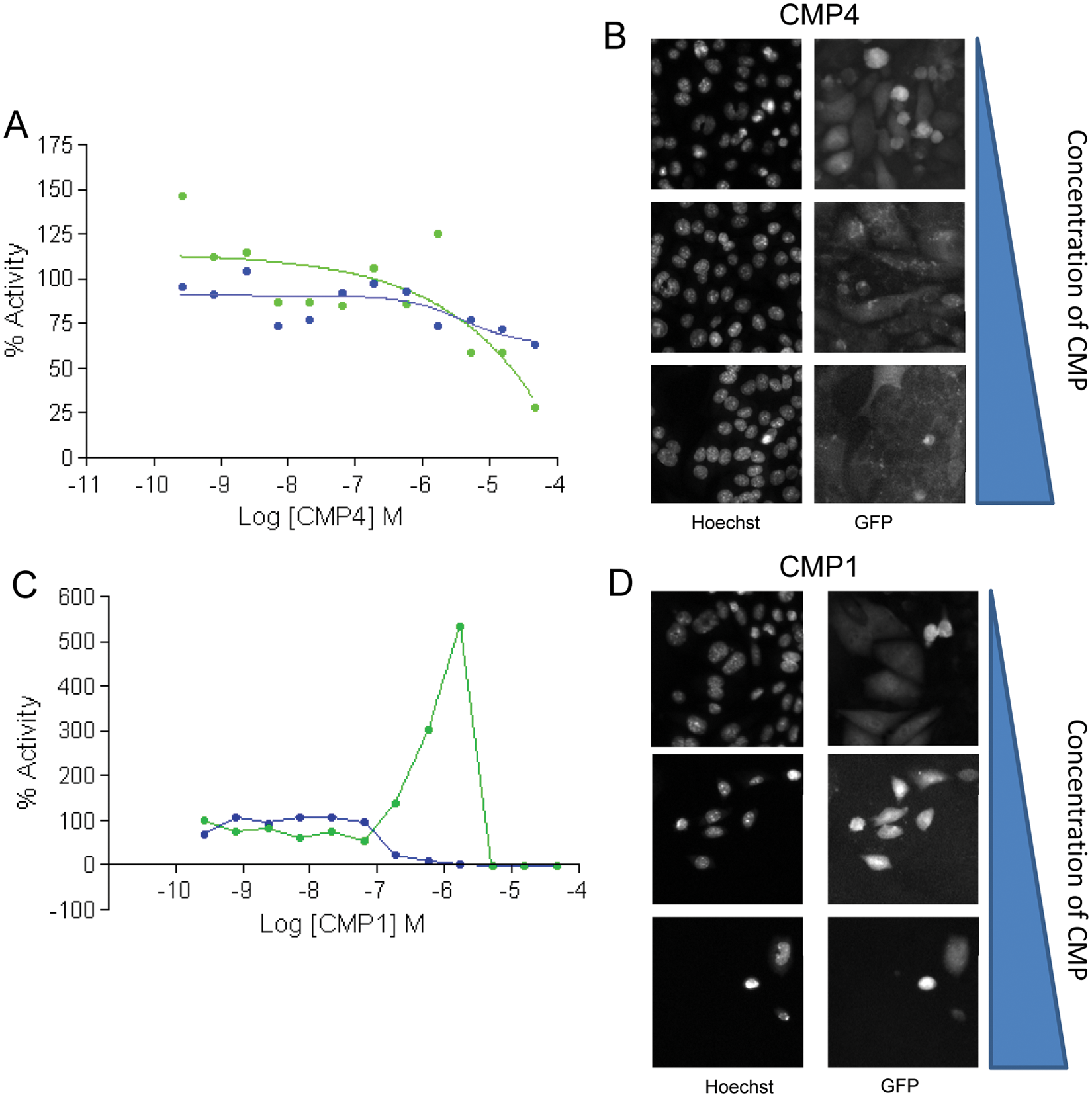
Results from the 4T1.2 Nanog-GFP reporting imaging assay. (
Discussion
Reports of the drug-resistant nature of CSCs prompted our lab to design assays that could be used to screen for compounds that inhibit the growth of these very aggressive cancer cells. We chose to investigate the CSC models of prostate cancer and pancreatic cancer because of the limited treatments currently available for advanced stages of these two diseases and their highly lethal metastatic nature. CSC populations in an HTS setting are challenging because of the difficulty to isolate and scale up cell production. As previously mentioned, these cells are commonly isolated using FACS for certain cell adhesion proteins such as CD44, CD24, or CD133. Adapting this method of CSC isolation to produce enough cells to conduct an HTS assay is not practical because of the cost of the antibodies and also the time required to sort the millions of cells needed. To date, because of these constraints, very few HTS assays have been performed using CSCs. A previously published study described an HTS assay that used CSCs generated from highly transformed cells called HMLER breast cancer cells. 23 These cells were transformed to have a mesenchymal phenotype (a hallmark of CSCs) by downregulation of E-cadherin, producing expression of high levels of CD44 and low levels of CD24. 23 This screen identified salinomycin as a CSC cytotoxic compound. Our approach was to generate CSCs using the spheroid technique and develop a cell growth assay in a 1536-well microplate format to screen for compounds that are cytotoxic against these highly aggressive CSCs. The spheroid technique affords for the production of large amounts of CSCs and therefore enables the availability of these cells for the use in HTS without the need to engineer the cells.
To validate the design of our CellTiter-Glo–based proliferation assay, we first examined the effects of salinomycin, the compound identified in the CSCs screen with HMLER cells. Previous studies have demonstrated that this compound is effective at inducing apoptosis in prostate cancer cells and interferes with the end-stage progression of hormone-indifferent and chemotherapy-resistant prostate cancer. 31 Importantly, nonmalignant RWPE-1 prostate cells were relatively less sensitive to salinomycin-induced lethality. 31 In pancreatic cancer, it was recently shown that in combination with the standard-of-care gemcitabine, which targets non-CSCs, salinomycin inhibited the growth of pancreatic cancer cells in vitro and in vivo by inhibiting the growth of CD133+ or side-population isolated CSCs. 32 In both of our CSC models, salinomycin was able to effectively, reproducibly, and significantly inhibit their growth in a 1536-well microplate format. A second positive control that we employed was the protease inhibitor bortezomib based on personal communication that it was quite effective at inhibiting the growth of mouse breast CSCs (unpublished results from Thomas Sayers Laboratory). In both systems, bortezomib was very effective at inhibiting growth of not only the adherent cells but also the spheroid-derived populations. Unfortunately, although bortezomib appears in vitro as a very promising treatment for pancreatic cancer, when tested in vivo in a mouse orthotopic model of pancreatic adenocarcinoma, it was ineffective, further demonstrating the need for new compounds that attack this deadly disease. 33
The 1536-microplate spheroid-derived CSC growth assay was used to screen an in-house small-molecule collection consisting of 112 compounds targeting oncology-relevant pathways/mechanisms of survival, also referred to as the Mechanism Interrogation PlatE or MIPE plate. This library includes approved chemotherapeutics, preclinical candidates, compounds in different phases of clinical development, and investigational compounds targeting key pathways and mechanism in oncology. This library includes known drugs with a long history of clinical use such as 5-fluorouracil and vincristine, as well as molecules in advanced clinical assessments that embody the contemporary approach of highly targeted enzyme and pathway inhibitors such as amuvatinib (DNA repair inhibitor) and talmapimod (p38 MAPK inhibitor). Because drug combinations can offer a better chance at providing high efficacy while reducing dose and potentially toxicity, molecules that failed in various phases of trials such as tozasertib (Aurora kinase inhibitor) and alvespimycin (HSP90 inhibitor) are also included. Recently disclosed clinical compounds targeting novel cellular targets relevant to the cancer types, including inhibitors of spleen tyrosine kinase (SYK), Bruton’s tyrosine kinase (BTK), PKCγ, MEK, HSP90, PARP, and PI(3)K, are also included in the collection. In addition, the library includes a degree of chemical diversity for each target to be able to explore compound activity differences associated with varying selectivity profiles. For example, many epidermal growth factor receptor (EGFR) inhibitors are included—gefitinib, lapatinib, neratinib, and dacomitinib—to help determine target versus compound-based activity. The composition of this collection allows us to potentially find drugs that can be efficiently used to treat CSCs, as well as to find biological information on what pathways are relevant for the survival of CSCs.
The results from the screen of this collection suggest that the major driver of cellular survival to the compounds tested is not necessarily the differentiation state of the cells but rather the cancer type. Although there might be a small trend toward CSCs being more resistant than the parental adherent cells, which are more differentiated, in general, the difference in compounds’ cytotoxic effect between LNCaP and PANC1 cells is larger than between spheres and adherent cells. However, two compounds were identified using the % viability criteria that inhibited growth of both spheres and adherent cells from both the LNCaP and PANC1 cells (
When applying the CRC hit selection method, five compounds where shown to be pan-active against both cell lines and growth conditions, with only one compound overlapping between the two analysis methodologies, CMP2 (
The MIPE compounds were also tested for their ability to regulate the stem cell state in CSCs as a potential mechanism for compounds to reduce resistance of these cells to cytotoxic agents. The Nanog gene regulates the stem cell state in embryonic stem cells and has been recently identified as a key regulator of CSCs in many systems, including prostate, pancreas, and breast.26–30 These cells were originally derived from a spontaneous metastatic carcinoma found in a BALB/cfC2H mouse and termed the 4T1 cell line. 34 Further single-cell cloning led to the development of a highly aggressive line, 4T1.2, which metastasizes to bone, lungs, and other sites in the animal. The 4T1 model was also used in the aforementioned HMLER screen to confirm effective inhibition of metastasis formation with the hits identified in their screen. Using a highly aggressive mouse breast cancer cell line 4T1.2 that has been stably infected with a Nanog-GFP reporter as a model system allowed us to screen the MIPE library with high-content technology in addition to the luminescent cell proliferation assay we developed. Unlike the prostate and pancreatic models of CSCs, this model is well established to visualize the effects compounds have on primary tumor formation as well as the appearance of metastases34,35 and is primarily why we selected it to screen using high-content imaging. Screening using a mixed population of cells is inherently difficult, and the majority of the data demonstrated that compounds were cytotoxic to these cells, positive or negative for GFP, as measured by reduced cell number. As mentioned above, this bias could be attributed to the fact that the library we screened contains oncology-based compounds that mainly target cancer cells by inducing cytotoxicity. Screening with additional libraries may remove this bias when using a high-content approach. Interestingly, for the top pan-active compounds selected from the cell proliferation assay, the mean nuclear Nanog-GFP signal per cell actually increased at high doses before no signal could be detected at those very high doses where there were no surviving cells in the well. Figure 5C , D shows the effect of CMP1 on the Nanog-GFP imaging assay. The images show how cells surviving treatment with CMP1 had very high Nanog expression. The heterogeneity of the 4T1.2-Nanog cells and types of compounds we are looking for is not well suited for an HTS assay using high-content imaging. However, by having both sets of data from the cytotoxicity assay and Nanog-GFP assay, one can begin to appreciate the value of the high-content set and the additional information it can provide about the biology of certain compounds. Finally, a few compounds actually increased the expression of Nanog without affecting nuclear count, and they will be followed up as potential CSC activators in our lab.
In conclusion, the research presented here demonstrates one of the first qHTS-based assays using true, nonengineered CSCs. The cell growth assay developed is a very sensitive and can be used to screen additional libraries with different types of CSCs in the future. In addition, we tested the MIPE library against mouse breast cells stably expressing a marker of stemness, Nanog. This assay proved to be quite heterogeneous and, similar to the proliferation assay, found hits resulting in cytotoxicity using the Hoechst nuclear staining channel irrespective of Nanog expression. Although this article focuses on selection of pan-active compounds, we are also pursuing compounds that are selective for cells from each cancer type. We are also planning to screen this cell proliferation assay with additional libraries of compounds as single agents and in combinations to determine which pathways need to be inhibited to produce complete toxicity against CSCs. We believe this will result in the development of novel therapies for aggressive forms of cancer and will specifically result in targeting metastasis and recurrence.
Footnotes
Acknowledgements
We thank Matthew Boxer, Damien Duveau, Christopher Leclair, and Jian-kang Jiang of NCATS for their assistance with acquiring the compounds for the MIPE library.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article confirmed statement is accurate.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was partly supported by the NIH Common Fund Molecular Libraries and Imaging Program, grant U54 MH084681. In addition, this project has been funded in part with federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.