
Abstract
Using fluorescence resonance energy transfer (FRET), we performed a high-throughput screen (HTS) in a reconstituted membrane system, seeking compounds that reverse inhibition of sarcoplasmic reticulum Ca-ATPase (SERCA) by its cardiac regulator, phospholamban (PLB). Such compounds have long been sought to correct aberrant Ca2+ regulation in heart failure. Donor-SERCA was reconstituted in phospholipid membranes with or without acceptor-PLB, and FRET was measured in a steady-state fluorescence microplate reader. A 20 000-compound library was tested in duplicate. Compounds that decreased FRET by more than three standard deviations were considered hits. From 43 hits (0.2%), 31 (72%) were found to be false-positives upon more thorough FRET testing. The remaining 12 hits were tested in assays of Ca-ATPase activity, and six of these activated SERCA significantly, by as much as 60%, and several also enhanced cardiomyocyte contractility. These compounds directly activated SERCA from heart and other tissues. These results validate our FRET approach and set the stage for medicinal chemistry and preclinical testing. We were concerned about the high rate of false-positives, resulting from the low precision of steady-state fluorescence. Preliminary studies with a novel fluorescence lifetime plate reader show 20-fold higher precision. This instrument can dramatically increase the quality of future HTS.
Introduction
The sarcoplasmic reticulum calcium ATPase (SERCA) is responsible for clearing Ca2+ from the sarcoplasm, leading to muscle relaxation. In the heart, and particularly in the ventricular myocardium, SERCA2a is under tight inhibitory regulation by phospholamban (PLB), a small (52-residue) single-pass transmembrane protein that is co-expressed with SERCA in the sarcoplasmic reticulum (SR). PLB decreases the apparent Ca2+ affinity of SERCA, thus inhibiting Ca2+ pumping at diastolic (submicromolar) concentrations of intracellular [Ca2+]. 1 The transmembrane domain of PLB is responsible for the inhibition of SERCA, 2 which is relieved by systolic (micromolar) [Ca2+] or when PLB is phosphorylated at residues 16 and/or 17 in the cytoplasmic domain. 3 This enables the myocardium to tap into a Ca2+ pumping reserve necessary for the fight-or-flight response.
SERCA activity decreases significantly in heart failure (HF), resulting in incomplete and slower relaxation after each heartbeat, contributing to the altered calcium homeostasis that is a hallmark of HF. 4 The ratio of PLB to SERCA is increased in HF, thus resulting in increased SERCA inhibition. Rescuing SERCA activity in the heart has been achieved via gene therapy, overexpressing SERCA2a or expressing noninhibitory PLB mutants. 5 SERCA activation has been shown to be harmless in normal animals and to improve muscle function in animal models of several types of heart disease, as well as muscular dystrophy. 5 The SERCA2a gene therapy treatment has been shown to be effective for human HF patients in phase 2 clinical trials. 6 These results clearly validate SERCA activation as a target for HF therapy. However, given the inherent complexity of gene therapy, we are pursuing an alternative approach—the direct activation of SERCA by small-molecule drugs—which would enable acute hospital intervention in HF as well as chronic use in case of reduced cardiac activity.
Industrial and academic researchers have searched intensively but unsuccessfully for compounds that dissociate PLB from SERCA to relieve inhibition of Ca2+ transport specifically in the heart and improve the prognosis in HF patients. 7 Based on our basic research on the SERCA-PLB system, it is likely that prior efforts failed primarily because of the lack of a robust high-throughput assay (HTS) for inhibitors of the SERCA-PLB interactions. Previous attempts to study this system used cumbersome functional assays that required several minutes each. 7 We use that kind of functional assay for our secondary screen, as discussed below, but successful HTS requires a primary assay that (1) detects directly a signal that is strongly correlated with function, preferably at molecular level, and (2) is truly high throughput, so that thousands of compounds can be tested in a day. We have approached both requirements using fluorescence resonance energy transfer (FRET). We developed an assay to measure the SERCA-PLB interaction directly with FRET, using purified SERCA and PLB labeled with fluorescent dyes in reconstituted membranes ( Fig. 1 ). 8 In this assay, an excited donor on SERCA transfers energy to a nearby acceptor on PLB, thus decreasing the donor’s fluorescence intensity ( Fig. 1 ). Using this assay, we discovered that relief of SERCA inhibition does not require dissociation of the SERCA-PLB complex ( Fig. 1 , right), as proposed by others. 9 Instead, a subtle allosteric structural change of the SERCA-PLB complex, without dissociation, is sufficient to relieve inhibition. ( Fig. 1 , left).8,10,11 Thus, the FRET signal should detect any structural change within SERCA that alters the distance from the SERCA-bound donor to the acceptor on PLB.
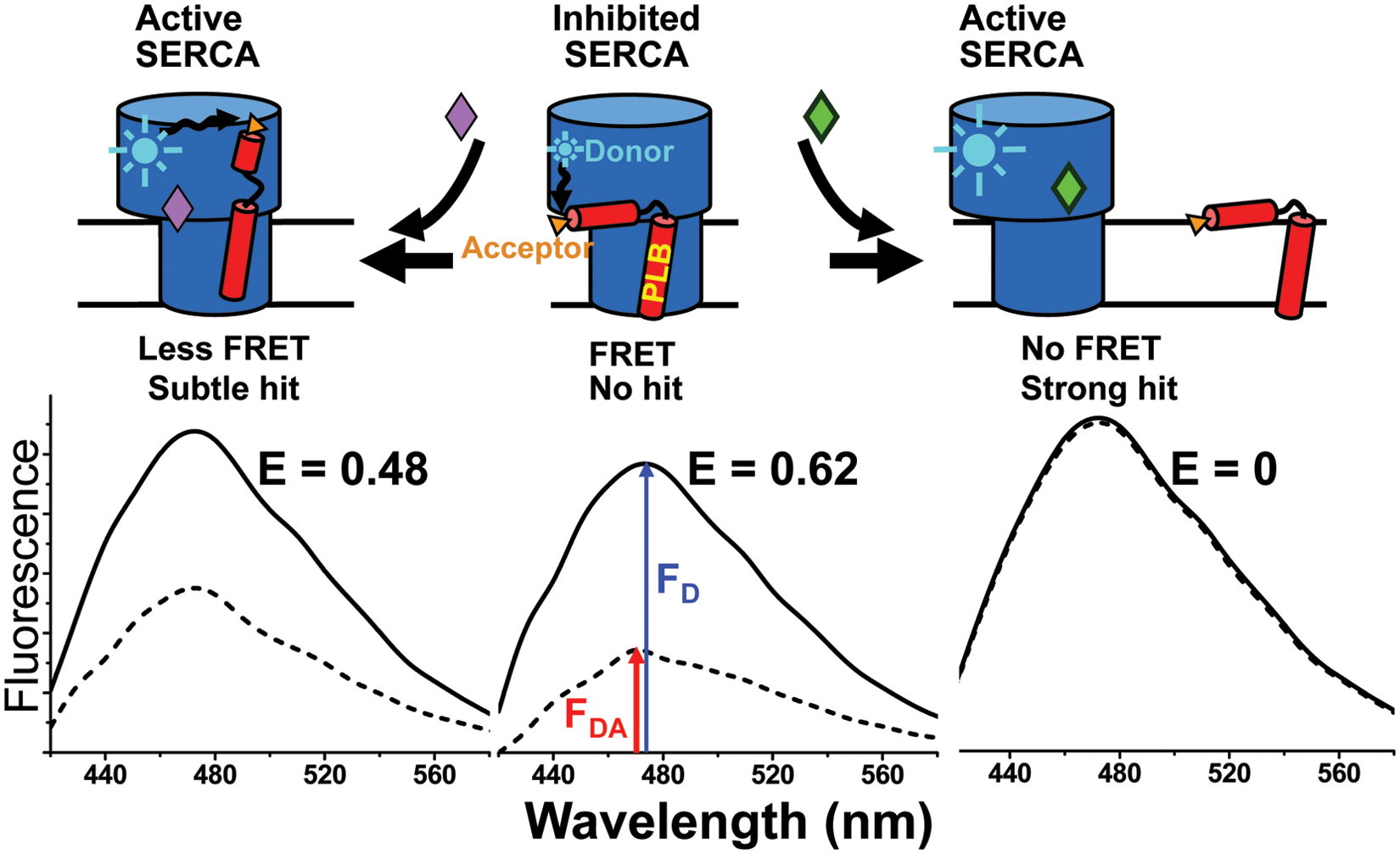
Fluorescence resonance energy transfer (FRET) assay for disruption of the inhibitory interaction between sarcoplasmic reticulum Ca-ATPase (SERCA) and phospholamban (PLB). (
Fortunately, because of the R-6 distance dependence of FRET, it can detect subtle structural changes resulting from protein-protein interactions and/or conformational changes. In addition, a precise FRET measurement can be done in less than a second, offering the clear potential for a high-throughput assay. Here we report results from an HTS we have conducted using our SERCA-PLB FRET assay ( Fig. 1 ), 8 modified for a fluorescence microplate reader. We screened a small-molecule library for compounds that disrupt the physical interaction between donor-labeled SERCA and acceptor-labeled PLB, aiming to increase the Ca2+ transport activity of SERCA by disinhibiting the enzyme. It was expected that most compounds would not change the FRET efficiency E ( Fig. 1 , center). If the SERCA-PLB complex is dissociated, FRET should be completely eliminated ( Fig. 1 , right). But if a more subtle structural change occurred, without dissociation ( Fig. 1 , left), there would be a smaller change in FRET. This screen was specifically targeted at the SERCA-PLB interface with two main goals: (1) reversing inhibition of SERCA by PLB and (2) thus specifically targeting SERCA in the heart.
Materials and Methods
The 20 000-compound DIVERSet library of molecules of less than 600 molecular weight was obtained from ChemBridge (San Diego, CA). The screen was conducted in NUNC 242764 384-well black-wall/clear-bottom microplates (Nalge Nunc International, Rochester, NY); CappAero 16-channel pipettes were from Capp A/S (Odense, Denmark). The Gemini EM microplate fluorimeter and Aquamax DW4 liquid dispenser were purchased from Molecular Devices (Sunnyvale, CA). Fluorophores 5-((((2-iodoacetyl)amino)ethyl)amino)naphthalene-1-sulfonic acid (IAEDANS) and 4-((4-(dimethylamino)phenyl)azo)benzoic acid, succinimidyl ester (DABCYL) were purchased from Invitrogen.
Isolation of Sarcoplasmic Reticulum Vesicles
Skeletal muscle SR membrane vesicles were isolated from longissimus dorsi obtained from New Zealand white rabbits, as previously described. 8 Cardiac SR membrane vesicles were isolated from ventricular myocardium obtained from fresh pig hearts. 12
SERCA Preparation and Labeling
SERCA was purified by Reactive-Red (Sigma) affinity chromatography from skeletal SR using a procedure described previously 8 and was stored at −80 °C until further usage. Purified SERCA was labeled with IAEDANS, a small fluorescent probe that reacts at Cys-674 in the P-domain, as described previously. 8 Samples of AEDANS-SERCA were flash-frozen and stored in the dark at −80 °C until further usage.
PLB Synthesis and Labeling
PLB (canine sequence) was assembled on Fmoc-Leu-PEG-PS resin by Fmoc chemistry using a PE Biosystems Pioneer peptide synthesis system, as previously reported. 8 The N-terminal amino group of unlabeled PLB was acetylated using acetic anhydride. For FRET, the nonfluorescent acceptor DABCYL was reacted at Lys-3 of WT-PLB (denoted DABCYL-PLB). The composition and concentration of synthetic PLBs were confirmed by MALDI-TOF and amino acid analysis, and samples were stored in methanol at −20 °C.
Co-reconstitution of SERCA and PLB
Samples were prepared fresh daily, scaling up our previous method, 8 to 2 mg reconstituted AEDANS-SERCA (donor-only sample) and 2 mg AEDANS-SERCA co-reconstituted with 530 µg DABCYL-PLB (to obtain a donor + acceptor sample with a molar PLB/SERCA = 5, close to that found in the normal heart). 13 The molar ratio of lipid/SERCA was 700. These amounts are sufficient for five donor-only and five donor + acceptor 384-well plates, plus one quality-control test plate.
Plates and Plate Preparation
The NUNC plates were chosen for their relatively small and uniform intrinsic fluorescence signal in the 420 to 600 nm wavelength range. The DIVERSet library was diluted in DMSO and reformatted for the screen in these plates at the University of Minnesota High-Throughput Biological Analysis Facility. Columns 1 and 24 of each plate were reserved for no-compound controls (20/plate) and buffer blanks (12/plate); these wells contained the same volume of DMSO as the wells containing library compounds. Columns 2 to 23 contained library compounds in duplicate. These plates were sealed and stored at −20 °C until used in the screen. On the day they were used in the screen, the assay plates were equilibrated to room temperature (25 °C), then spun 5 min at 1000 × g in an Eppendorf 5810R centrifuge equipped with an A-4-81 rotor and microplate adaptor buckets. Sample containing 90 nM AEDANS-SERCA (78 µL) was applied to the assay plate over the 2 µL of test compound using an Aquamax DW4 liquid dispenser (Molecular Devices) to obtain a final compound concentration of 10 µM. Before reading, assay plates were incubated for 20 min at room temperature.
Fluorescence Data Acquisition
Typically, in HTS assays using fluorescence intensity, a single-wavelength measurement is recorded for each well. In this screen, the relatively low brightness of AEDANS-SERCA and high probability of spectral distortion (e.g., due to test-compound fluorescence), particularly at wavelengths shorter than 500 nm, led us to acquire full fluorescence intensity spectra for each well. This increased amount of information aided in determining whether a reading was reliable or skewed by compound fluorescence or by other factors contaminating the AEDANS-SERCA spectrum. Plates were read in a Gemini EM microplate fluorometer (Molecular Devices) with excitation at 355 nm from a Xenon flash lamp (1 J/flash) and a 420 nm long-pass emission filter. Fluorescence emission spectra were recorded from 420 to 600 nm, with 10 nm step size.
Fluorescence lifetime (FLT) measurements were conducted in a prototype of the NovaFluor plate reader (Fluorescence Innovations, Bozeman, MT), which uses direct waveform recording to provide the rate of data acquisition necessary for HTS. 14 Fluorescence was excited 355 nm using a 10 kHz passively Q-switched microchip laser (JDS Uniphase). Fluorescence emission was focused into a photomultiplier tube module (Hamamatsu) and digitized with 0.2 ns resolution. This instrument can scan a 384-well plate in less than 2 min, yielding waveforms with S/N = 100 in each well for samples containing 90 nM AEDANS-SERCA. This rate of data acquisition at high S/N is at least 100 times faster than achievable by other FLT plate readers.
HTS Data Analysis
Fluorescence spectra were corrected by subtracting the signal corresponding to buffer controls within the plate. Compounds that distorted the fluorescence spectrum were excluded from the hit selection. Donor-only and donor-acceptor controls (i.e., containing no library compound, only the corresponding 2 µL volume of DMSO) were used to determine standard deviation (SD) within the plate. The FRET efficiency E was calculated according to:

where FD is the fluorescence intensity of the donor-only sample, which is decreased to FDA by the acceptor in the donor-acceptor sample. Assay quality was determined based on controls on each plate, as indexed by the z′ parameter 15 :
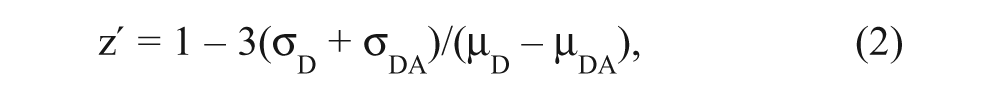
where σD and σDA are the SDs of the control FD and FDA, respectively, and µD and µDA are the means of the control FD and FDA, respectively. A compound was considered a hit if it decreased E by more than three times the SD of the no-compound controls.
Secondary Functional Assays
Ca-ATPase Activity
The reconstituted samples used in HTS were characterized as a function of pCa using an NADH-linked, enzyme-coupled ATPase assay adapted for 96-well microplates ( Fig. 2 ). 16 Each well contained 2 µg (skeletal) or 7 µg (cardiac) of SR vesicles (adjusted for the different SERCA contents of skeletal and cardiac SR), 50 mM MOPS (pH 7.0), 100 mM KCl, 5 mM MgCl2, 1 mM EGTA, 0.2 mM NADH, 1 mM phosphoenol pyruvate, 5 IU of pyruvate kinase, 5 IU of lactate dehydrogenase, 3.5 µg/mL of the calcium ionophore A23187, and CaCl2 added to set free [Ca2+] to the desired values. The assay was started upon the addition of ATP at a final concentration of 5 mM and read in a SpectraMax Plus microplate spectrophotometer. The Ca-ATPase assays were conducted over a range of [Ca2+], and the ATPase activities were fitted using the Hill function
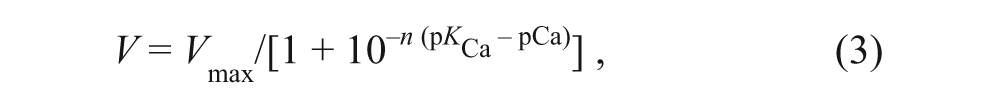
where V is the initial ATPase rate, Vmax is the ATPase at saturating [Ca2+], n is the Hill coefficient, and pKCa is the apparent Ca2+ dissociation constant. The assay provides a confirmation of the functional integrity of SERCA reconstituted in the absence of PLB ( Fig. 2 , blue), and the right-shift in the calcium curve when co-reconstituted with PLB ( Fig. 2 , black) was diagnostic of the correct functional coupling within the SERCA-PLB complex. We used this method as our primary orthogonal assay to determine whether a hit compound, identified by the FRET screen, activated SERCA function. A compound that reverses the inhibitory effect of PLB on SERCA (e.g., by completely dissociating the complex) should shift the Ca curve to the left ( Fig. 2 , black to blue), thus resulting in an increased rate of Ca2+ transport at nM [Ca2+] (as in diastole). However, activation can also be achieved by increasing Vmax ( Fig. 2 , black to red).
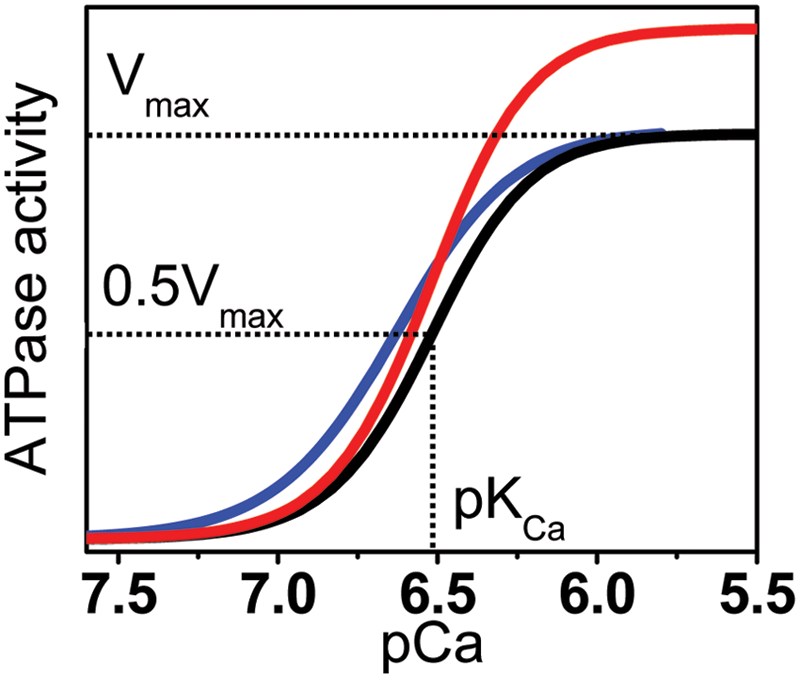
Functional effects of a hit. Desirable compounds may act on the Ca2+ dependence of sarcoplasmic reticulum Ca-ATPase’s ATPase activity (black) to increase its apparent Ca2+ affinity (pKCa, blue) or to increase the maximum rate (Vmax, red), or a combination of both (not shown). Curves represent fits of equation 3 to typical Ca-ATPase data.
Cardiomyocyte Assays
Adult male Sprague-Dawley rats weighing 250 to 300 g were obtained from Charles River Laboratories and used for the assessment of myocyte contractility. Cardiac myocytes were obtained by enzymatic isolation as described previously. 17 Left ventricular myocytes were suspended in a buffer containing 10 mM HEPES (pH 7.4), 131 mM NaCl, 4 mM KCl, 2 mM MgCl2, 1 mM CaCl2, and 10 mM glucose. Cells were incubated with either test compound dissolved in DMSO or DMSO vehicle only for at least 10 min before the measurements of cellular contractility. Myocyte contractility was measured using the IonOptix video-based edge detection system (IonOptix Corporation Milton, MA). 18 Sarcomere length was monitored at 25 °C in field-stimulated myocytes at 1 Hz, with 4 ms pulse duration, 30 V using a STIM-AT thermostated stimulator.
Measurement of [Ca2+]i transients was carried out as described previously. 19 Myocytes were loaded with the membrane-permeant fluorescent Ca2+ indicator fura-2 AM (Invitrogen). After myocyte incubation with the indicator for 15 min at 25 °C, fluorescence imaging was conducted using the IonOptix photomultiplier system, with excitation from a 75 W halogen lamp for 0.5 s at 360 nm then at 380 nm for the duration of the recording protocol. Emission was recorded at 500 nm (40 nm bandwidth). Cells were observed using an inverted microscope through an Olympus Fluor 40× oil objective. We measured multiple, randomly chosen myocytes treated with test compound or DMSO vehicle.
Results
HTS Performance
Tests were conducted with control samples to determine the precision of our assay. The relatively weak IAEDANS fluorescence spectrum is prone to distortion by test- compound fluorescence or by other factors contributing to sample irregularity (e.g., bubbles, particulates, variations in plate background). This could make the FRET measurement imprecise, thus negatively affecting the screen quality. Therefore, we devised methods to minimize spectral distortions during data acquisition and to identify distorted spectra during data analysis. Use of a nonfluorescent acceptor (DABCYL) facilitates distortion detection and correction because it allows using the entire donor fluorescence spectrum for FRET calculations. In preliminary assessments of the assay quality, we found that spectral distortions were less at longer wavelengths of the IAEDANS spectrum (>500 nm). For AEDANS-SERCA samples, we found that the coefficient of variance (CV = σ/µ × 100%) was significantly higher at 470 nm (10%) than at 520 nm (6%). We measured approximately the same CV for the donor-only sample and for samples in which FRET efficiency E was about 0.5. To predict assay quality, we calculated the factor z′ based on CV and signal window (ΔE), with equation 2 rewritten as
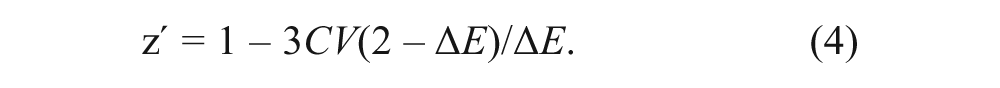
Based on the measured CV and on the average FRET efficiency of the 5DABCYL-PLB/AEDANS-SERCA samples (E = 0.57 ± 0.09), we predicted z′ parameters of 0.25 ± 0.22 and 0.54 ± 0.11 for readings at 470 and 520 nm, respectively ( Fig. 3A ). Therefore, the screen was preliminarily deemed at least doable (with readings at 470 nm) and excellent (with readings at 520 nm) if complete inhibition of FRET serves as the condition for hit selection.
High-throughput screen results. Compounds were screened, in duplicate, at a concentration of 10 µM (in DMSO). (
The library was screened in 110 duplicate plates, and the apparent effect of compounds on the FRET efficiency, ΔE, is shown for all compounds in Figure 3B , with positive and negative ΔE values indicating increase or decrease, respectively, relative to the E of controls. Our goal was to identify compounds that significantly decrease the FRET efficiency. The distribution of ΔE can be reasonably fitted by a Gaussian, centered at approximately the control value, µ = 0.5% ± 0.3%, with a global SD, σ = 8% ( Fig. 3C ). To decrease the probability of false hits, all compounds were screened in duplicate on the same plate. Z′ was calculated for each plate based on the no-compound controls (20/plate). For the actual screen, the average daily z′ value was 0.4 ± 0.2 (n = 22).
A slight asymmetry in the distribution is observed ( Fig. 3C ), with negative ΔE values somewhat more numerous than positive values. This is due to compounds that have a fluorescence spectrum comparable in intensity over the same wavelength range as the spectrum of AEDANS-SERCA. Such compounds increase the total fluorescence and thus will appear to decrease FRET. The compounds distorting AEDANS-SERCA spectra (donor-only) were identified based on their effect on the donor-only spectrum and were eliminated from consideration during the hit selection process.
Only data from plates with z′ ≥ 0 were analyzed for hits. Although a significant number of compounds increased ΔE beyond the 3σ threshold ( Fig. 3B ), most of these values appear to result from irregularities in the fluorescence spectra, so the corresponding compounds were filtered out. Our final set of hits consisted of 43 compounds, about a 0.2% hit rate (which is considered low for most screens but is typical for screens of protein-protein interaction), which we ranked based on the observed E value relative to controls (E0, no compound; Fig. 3D ). However, when these 43 preliminary hits were rescreened exhaustively (n ≥ 10) at 10 µM concentration, only 12 (28%) were found to cause a significant decrease in FRET (beyond the 3σ threshold). These 12 compounds were selected for secondary functional screens.
Secondary Assays
Ca-ATPase Activity in Purified SR
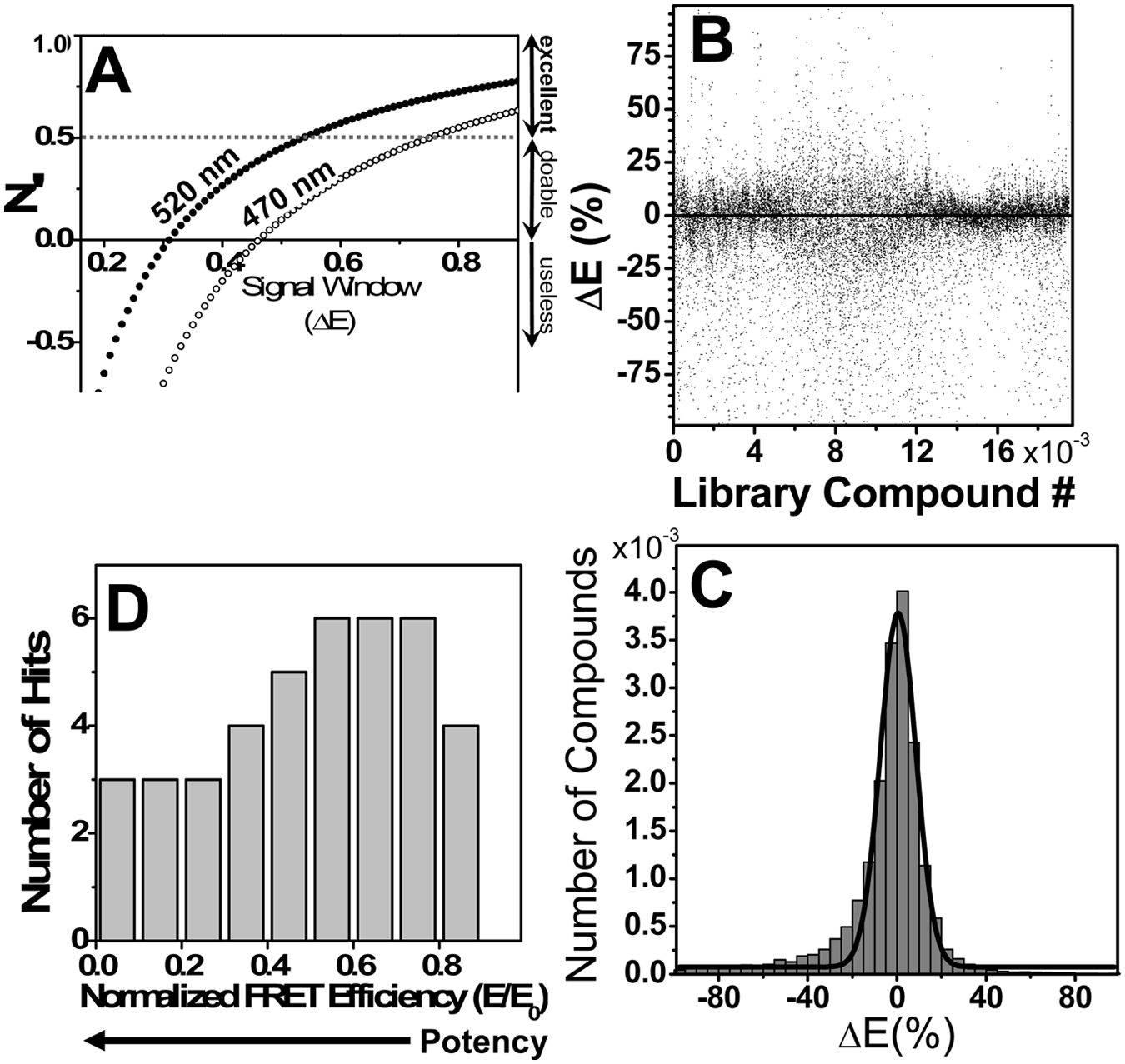
The functional effects of the hit compounds were first measured in vitro using Ca-ATPase assays, which were carried out in skeletal SR (SERCA1 only) and cardiac SR (SERCA2a and PLB) ( Fig. 4 ). We were searching for either of the effects expected to be useful in the treatment of HF, which are illustrated in Figure 2 . Based on the design of the assay (i.e., for detection of disruptions of the SERCA-PLB interaction), we expected that the main effect of the hits would be a left shift in the Ca-curves, as depicted in Figure 2 (blue vs. black curve), corresponding to an increase in the apparent Ca-affinity of SERCA due to dissociation from PLB. Surprisingly, when we measured the effects of the 10 µM hit compound on the Ca dependence of SERCA’s ATPase activity, we found that none of the tested compounds caused a significant shift in the apparent Ca affinity. However, 6 of the 12 compounds tested at 10 µM increased Vmax by at least 10% in both cardiac and skeletal SR (red vs. black curve in Fig. 2 ). Results from the two most effective of these compounds are shown in Figure 4 . CDN 1033 increased Vmax by about 50% in cardiac SR and 65% in skeletal SR. None of the compounds had effects that were specific for cardiac SR. In fact, in several cases, the Vmax increase was greater in skeletal SR (where SERCA1a is expressed in the absence of PLB) than in cardiac SR (where SERCA2a is coexpressed with PLB; Fig. 4 ). Therefore, these results show that the hit compounds identified by our screen act directly on SERCA and that their action is not dependent on the SERCA-PLB interaction or on a specific SERCA isoform. The significant differences in compound potencies observed for skeletal and cardiac SR could be due to intrinsic differences in the SERCA isoforms or to the presence of PLB in cardiac SR. Further assays on purified SERCA isoforms will be needed to distinguish these possibilities. All six hit compounds were subjected to a battery of other tests to examine their selectivity for SERCA, assessing their effects on numerous other ion pumps and channels (e.g., Na/K-ATPase, L-type Ca channel, ryanodine receptor), and no significant functional effects were observed. Thus, these compounds are the first reported small-molecule activators that are specific for SERCA. Results from the dose response of Vmax indicate EC50s in the micromolar range for both CDN1001 and CDN1033 for both cardiac and skeletal SR ( Fig. 4C , D ). The physical properties of CDN1001 and CDN1033—molecular weights less than 400, log P values less than 4, and polar surface areas of less than 100—suggest they would be viable lead candidates for optimization toward clinical applications. Medicinal chemistry efforts are in progress to increase the affinities of these compounds for SERCA by generating compound libraries inspired by the structures of the hits.
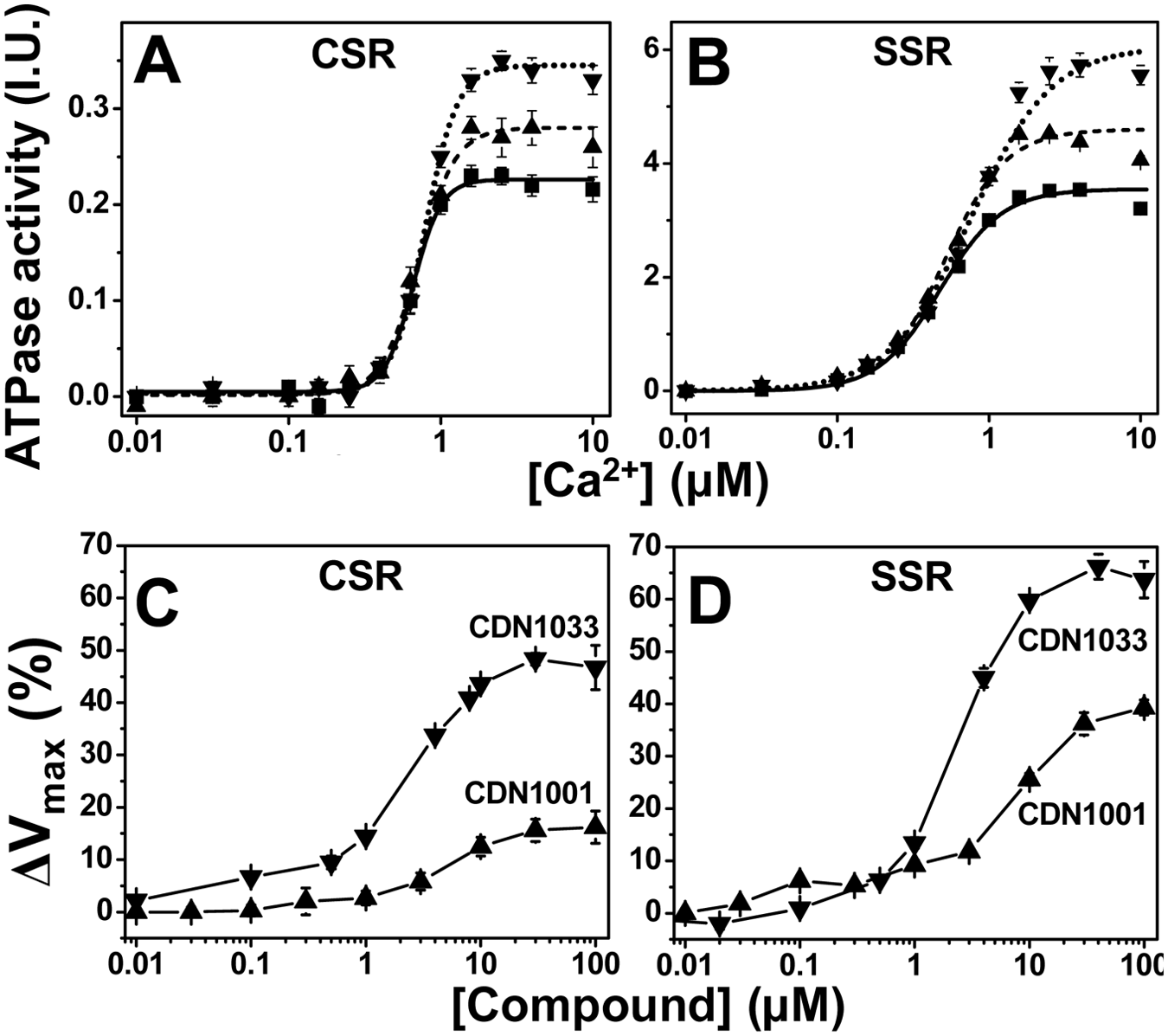
ATPase assays. ATPase activity was measured after 20 min incubation in the presence of either CDN1001 (up triangles) or CDN1033 (down triangles) or DMSO control (squares). Top (
Cardiomyocyte assays
Myocyte contractility assays were performed to determine whether the hit compounds that were confirmed by the ATPase assays have effects on cells isolated from the tissue meant to be targeted by the drug that will be developed eventually based on these compounds. We used myocytes isolated from rat left ventricular myocardium. The sarcomere length of field-stimulated (1 Hz) myocytes was monitored over time, measuring both length and Ca2+ transients ( Fig. 5 ). Four compounds significantly increased contractility, as measured by sarcomere shortening ( Fig. 5A ). The effect of CDN1001 in enhancing the amplitude of Ca2+ transients ( Fig. 5B , C ) is consistent with its effect on myocyte contractility ( Fig. 5A ), as well as on the Ca-ATPase activity of isolated cardiac SR ( Fig. 4 ).
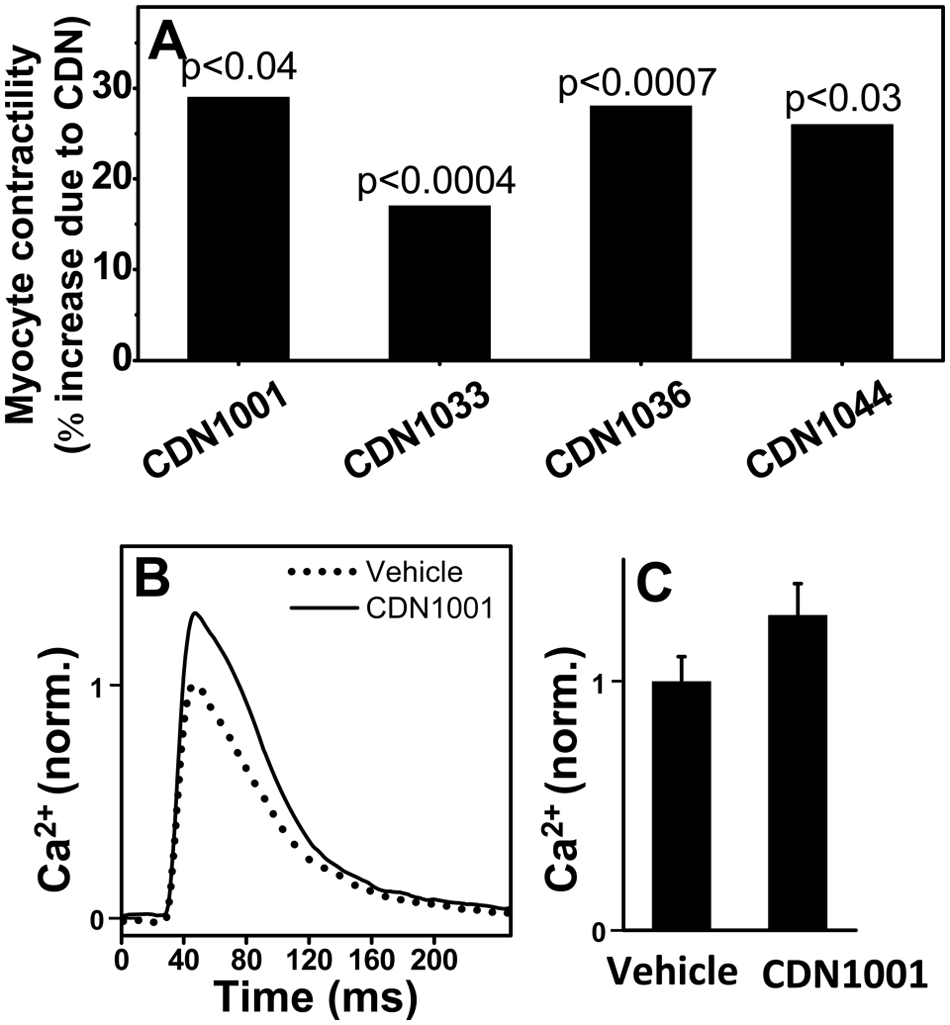
Effects of hit compounds on cardiomyocyte contractility and Ca2+ transients. (
FLT Plate Reader
At the start of the present HTS campaign, steady-state fluorescence intensity detection was the available technology in fluorescence microplate readers. A major factor limiting effectiveness of HTS is inadequate precision provided by conventional, steady-state fluorescence plate readers, resulting in too many false-positives (wasting subsequent resources) and false-negatives (missing potential hits). 20 The precision of steady-state fluorescence intensity detection is highly dependent on the amount (concentration) of fluorophore in the well, which depends primarily on the precision of sample dispensing. These are the major factors that affected the quality of our HTS assay, as discussed above in the Results section ( Figure 3 ).
FLT detection, resolving the nanosecond emission kinetics following a laser pulse, provides a signal (time-resolved waveform shape) that is not affected by fluorophore concentration, so FLT offers great potential for increasing precision in HTS. Nevertheless, most fluorescence experiments, including FRET, are currently detected by intensity, primarily because the conventional FLT method, single-photon counting, is slow, requiring many seconds for a single measurement that yields adequate S/N (≥100) for high resolution and precision. We have recently solved this problem by using a FLT plate reader (Fluorescence Innovations, Minneapolis, MN) that uses direct waveform recording 14 to acquire accurate and precise subnanosecond-resolved fluorescence waveforms several times per millisecond. This unprecedented combination of speed and precision makes it possible to scan a 384-well plate in less than 2 min, acquiring high-quality FLT decays (S/N ≥ 100) of control samples containing 90 nM AEDANS-SERCA, as used in the original screen ( Fig. 6A ). When intensity is measured, we observe the well-to-well variability typical of fluorescence intensity plate readers (CV = 4.2%; Fig. 6B , gray). This is similar to the value we observed with the intensity plate reader (Molecular Devices Gemini) used in the original screen ( Fig. 3A ). However, when the mean FLT (calculated from the first moment of each decay) is measured, there is a dramatic increase in precision by a factor of 20 (CV = 0.2%). To overcome this difference in precision, it would be necessary to run 400 intensity measurements for each lifetime measurement. This improvement in precision is directly reflected in the predicted HTS assay quality, with z′ > 0.5 for changes in FRET efficiency as small as 0.07 ( Fig. 6C ). This sharp improvement in assay quality provides the sensitivity necessary to detect the subtle conformational changes we expect to be caused by most compounds that act as allosteric activators of SERCA and other drug targets.21–23
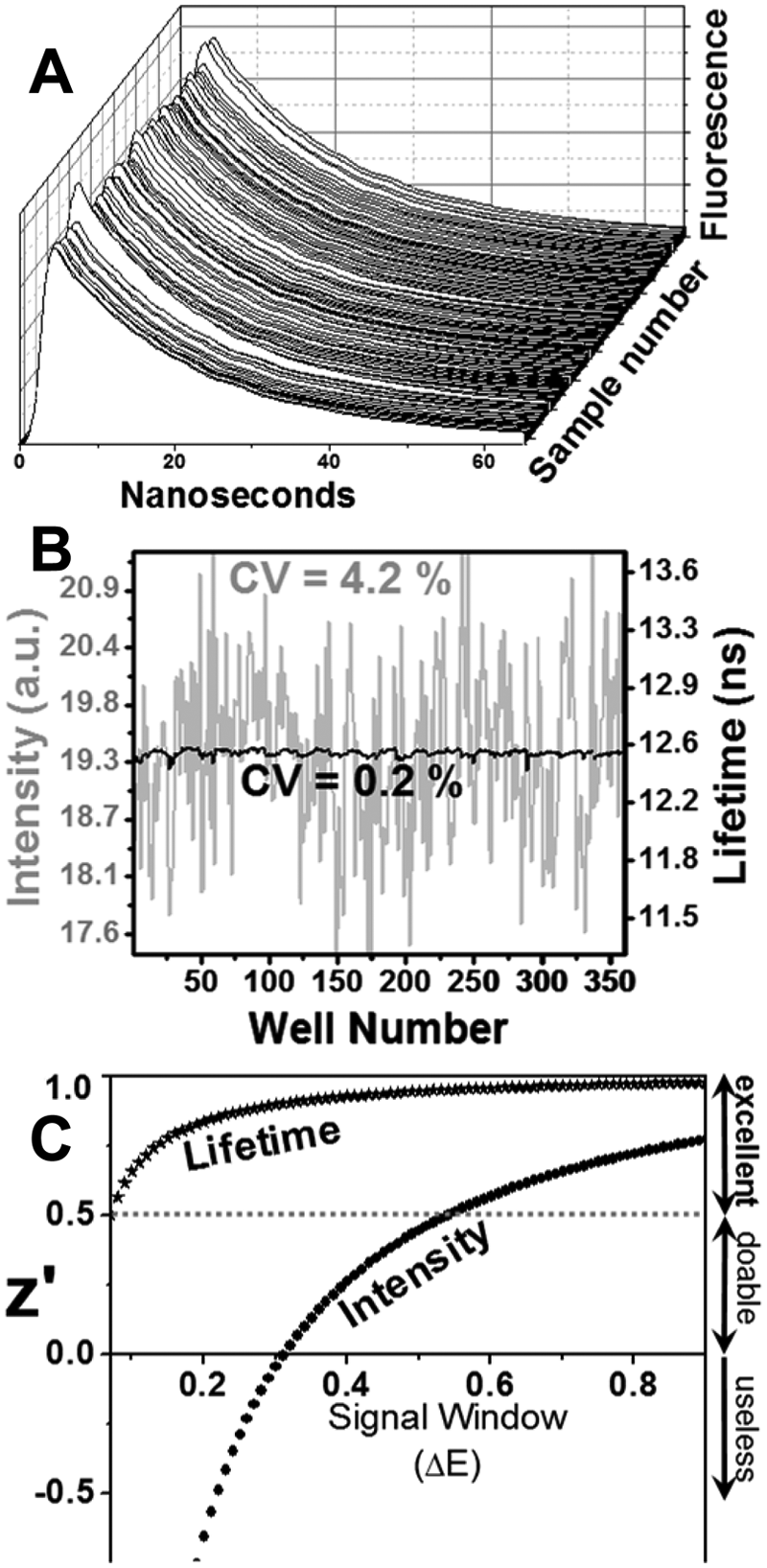
Screening with fluorescence lifetime (FLT) plate reader. (
Discussion
Deficient removal of Ca2+ from the cytoplasm of myocytes into the SR reservoir has been associated in human HF with reduced expression and activity of SERCA2a.24,25 To correct this situation, the rate of Ca2+ removal from the cytoplasm of failing cardiomyocytes must be enhanced. To achieve this goal, Hajjar and coworkers have validated a therapeutic approach (in HF animal models) consisting of SERCA overexpression in heart, where it restores normal Ca2+ cycling and cellular metabolism, leading to significant improvement in cardiac function.6,26–28 Indeed, the clear effectiveness and safety of AAV-SERCA gene therapy have recently been demonstrated in a phase 2 clinical trial for patients with advanced heart failure. 6 Although very promising, the gene therapy approach has limitations that leave a large percentage of the heart patient population outside its applicability. 28 Therefore, alternative means—pharmacological—of activating Ca2+ transport by SERCA should be identified.
To find compounds that activate SERCA, we conducted an HTS campaign using our proprietary high-throughput FRET assay, in which we detect energy transfer between a donor on affinity-purified SERCA and an acceptor on synthetic PLB. FRET is a popular spectroscopic method used for HTS of peptidase inhibitors and activators. However, in those assays, the peptide substrate is derivatized to carry both the donor and the acceptor. The use of FRET to identify modulators of protein-protein interactions, as we have done in this study, is more rarely used.
The donor-acceptor pair (IAEDANS-DABCYL) has a relatively short R0 (32 Å), 8 providing sensitivity to interprobe distance changes in a range that is expected to occur within the SERCA-PLB complex (16–48 Å). To our knowledge, this is the first HTS for modulators of protein-protein interactions of a reconstituted complex of membrane proteins. This approach was dictated by our initial goal: to develop an HTS assay that monitors the SERCA-PLB interaction interface in order to find compounds that disrupt the regulatory interaction between the two proteins or completely dissociate the complex. We found a very low hit rate (0.2% of the compounds screened), and 72% of these turned out to be false-positives. On the remaining 12 hits, we conducted secondary assays on the Ca-ATPase activity of SR membranes purified from cardiac and skeletal muscle. Half of these compounds were found to activate SERCA by at least 10%, but the effects were not specific for cardiac SERCA. These findings suggest that the compounds act directly on SERCA, probably via an allosteric mechanism to activate SERCA’s Ca-ATPase activity. These mechanistic details will be determined in future studies.
The compounds that activated SERCA in isolated SR samples were then tested on isolated cardiomyocytes to determine whether they can enhance cardiac contractility. About 50% of the hits significantly increased the amplitude of sarcomere shortening, which is an index of cardiomyocyte contractility. CDN1001 was studied in more detail, and we found that enhancement of sarcomere shortening by this compound was accompanied by a similar increase in the amplitude of the [Ca2+] transient, and both intracellular effects are similar in size with the effect of CDN1001 on SERCA Vmax. In light of these encouraging results with cardiac myocytes, CDN1001 has been further tested for effects on full heart and on animals. The results are quite encouraging but will be reported in a different article. Medicinal chemistry design and synthesis of CDN1033 analogs have led to a series of druglike allosteric SERCA agonists that show potential to reduce ER stress. These studies will be the focus of a different report.
Pharmaceutical companies have searched intensively for low-molecular-weight, systemically administered compounds that could relieve SERCA inhibition, improve Ca2+ homeostasis, and thus improve the prognosis in HF patients.7,29 They have failed for two principal reasons: (1) inadequate understanding of the structure-function correlations in the SERCA-PLB complex and (2) lack of an effective assay for detecting the SERCA structural changes. Our efforts thus far have led to the discovery of first-in-class small-molecule allosteric modulators of SERCA. This finding is significant because the only previously reported small-molecule SERCA activator, istaroxime, 30 is a molecule with several weaknesses precluding its clinical development, including polypharmacology, or lack of specificity (specifically, Na+, K+-ATPase inhibition); narrow therapeutic index; and reported toxicity. 31
Although the principle of our FRET HTS assay has been validated by secondary functional assays, showing that some FRET-detected compounds are potent SERCA activators, we were disappointed by the high rate of false-positives: 72% of the apparent hits from the initial FRET screen did not have a significant effect on FRET following repeated trials. This high rate of false-positives is clearly due to the modest precision of the intensity-based screen, and this lack of precision probably also implies a high rate of false-negatives: compounds having effects on FRET that are not detected (i.e., hits that are missed). This high rate of false-negatives greatly increases the size of the library that must be screened to obtain a given number of valid hits. Therefore, we have improved the signal-to-noise ratio of the assay by using the FLT plate reader. The data with this instrument in Figure 6 show clearly that FLT detection produces 20 times better precision on identical samples, changing the z′ value to >0.5 (i.e., excellent) even for very small changes in FRET (ΔE > 0.07).
We have carried out the first HTS campaign using reconstituted membrane proteins and a novel FRET assay that measured directly the interaction of SERCA and PLB. We identified several promising leads from a 20 000- compound library. None of these compounds were specific for SERCA in cardiac SR or for the SERCA-PLB interaction (as originally hoped), but several compounds in the primary FRET screen were found to activate SERCA selectively (i.e., without activating other ATPases or Ca2+ channels) in the micromolar concentration range. These are the first reports of such SERCA-specific activators. This success is particularly remarkable because of SERCA’s reputation as a difficult drug target. It is a difficult target in part because it is an integral membrane protein in complex with an integral membrane protein inhibitor (PLB) but mainly because the goal is enzyme activation. Drugs that activate enzymes are rare. The task of activating an enzyme is much more exacting than inhibiting it, most likely requiring subtle allosteric structural modulation, which places great demands on the quality of the HTS assay. For future HTS campaigns to identify allosteric activators, the FLT plate reader technology may be crucial due to its dramatic improvement in precision and assay quality ( Fig. 6 ). The compounds discovered in this and future SERCA-screening campaigns represent a promising new approach for treating heart failure, because ongoing gene therapy trials have already validated SERCA activation as an effective therapeutic strategy. 6 Evidence increasingly supports the proposal that activation of SERCA can be effective in treating other conditions, such as cancer 32 and diabetes 33 (SERCA2b) or muscular dystrophy34,35 (SERCA1a). Thus, compounds discovered in this screen are likely to have diverse therapeutic applications.
Footnotes
Acknowledgements
Christine Karim provided synthetic phospholamban; Nick Hahn and Mark von Keitz, at the University of Minnesota High Throughput Biological Analysis Facility, assisted with microplate formatting; and Bonnie Fedor provided excellent technical assistance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant to D.D.T. from the National Institutes of Health (GM27906), and by grant to D.D.T., R.L.C., D.J., and R.D. from Celladon Corporation. R.L.C., T.B., D.J., R.D., and D.D.T. were paid consultants for Celladon Corporation. K.M.Z. is president, director and CEO of Celladon Corporation. R.L.C., T.B., R.D., D.D.T., and K.M.Z. have been issued Celladon Corporation stock options. G.D.G. is president of Fluorescence Innovations, Inc.