
Abstract
A total of 149 human prostate tissues obtained from our institute were assessed: 52 specimens of benign prostate hyperplasia (BPH) and 97 specimens of prostate cancer (PCa). The methylation status of the genes of Adenomatous polyposis coli (APC) and glutathione-S-transferase-P1 (GSTP1) was analyzed by quantitative pyrosequencing. A methylation score (M score) was calculated to capture the combined methylation level of both genes. The methylation level of each single gene and that of both genes combined was significantly higher in PCa specimens than in BPH (each p < 0.001). The value of APC methylation, GSTP1 methylation, and M score for predicting PCa was measured by the area under the receiver operating characteristic (ROC) curve and reached 0.954, 0.942, and 0.983, respectively. The sensitivity and specificity of the M score in discriminating between PCa and BPH reached 92.8% and 100.0%, respectively. The M score was positively associated with the serum prostate-specific antigen (PSA) level (p trend < 0.001). Our study demonstrates that the quantitative measurement of two methylation markers might drastically improve the ability to discriminate PCa from BPH.
Keywords
Introduction
Prostate cancer (PCa) remains a major health issue and is one of the most frequent cancers among men. Early detection of cancer and subsequent treatment is usually associated with improved patient outcome compared with late-stage diagnosis. The decision to perform a biopsy is generally based on the results of serum prostate-specific antigen (PSA) testing and/or digital rectal examination. However, a significant proportion of biopsies are negative because of the low sensitivity and specificity of these tests. 1 Moreover, prostate biopsy can induce pain, discomfort, anxiety, and complications and increases medical costs. Thus, new tumor markers with better sensitivity and specificity for early cancer detection are needed.
DNA hypermethylation-induced silencing of tumor suppressor and DNA repair genes is a frequent phenomenon in cancer.2–4 The genes undergoing methylation during the early phases of tumorigenesis could potentially be used as markers for identifying individuals at increased risk of developing malignancy or for aiding in the diagnosis of early malignancy. Similarly, genes undergoing methylation during the progression of malignancy could potentially be used as prognostic markers.2–4 Although these hypermethylation markers are promising tools to detect cancer cells in tissues and body fluids, discordant results have been reported between aberrant DNA methylation detection methods.2–4 Therefore, new techniques are needed to provide reliable, sensitive, and fast results to study these potential biomarkers.2,3 Pyrosequencing (PSQ) is one of the most accurate methods available to quantify DNA methylation.4,5 It is a sensitive and highly reproducible method that is uniquely suited to the analysis of clinical specimens from which only small amounts of DNA can be isolated.5,6
Adenomatous polyposis coli (APC) and glutathione-S-transferase-P1 (GSTP1) are well-characterized tumor suppressor genes. Methylation of these genes is associated with PCa.5–12 Although a single molecular marker may be promising, the reliance on a single gene for the screening and diagnosis of PCa presents some limitations.10–12 These limitations could potentially be overcome by the simultaneous use of multiple sensitive and specific molecular markers. The aim of the present study was to assess the methylation of APC and GSTP1 using quantitative PSQ to determine whether APC and GSTP1 methylation may be associated with prostate clinicopathological parameters and to evaluate the relevance of APC and GSTP1 methylation in discriminating between benign prostate hyperplasia (BPH) and PCa.
Materials and Methods
Human Tissue Samples
A total of 149 human prostate tissues obtained from our institute were assessed (52 BPH and 97 PCa). Subjects with PCa underwent radical prostatectomy (RP) or palliative transurethral resection (TUR). Subjects with BPH underwent TUR. All tissues were macro-dissected within 15 minutes of surgical resection. Each prostate specimen was confirmed by pathological evaluation of fresh-frozen sections, and the rest of the tissue was frozen in liquid nitrogen and stored at −80 °C until use. The specimens were provided by the Chungbuk National University Hospital, a member of the National Biobank of Korea, which is supported by the Ministry of Health, Welfare and Family Affairs. The collection and analysis of all samples were approved by the Chungbuk National University Hospital Institutional Review Board, and informed consent was obtained from each subject.
DNA Extraction and Pyrosequencing Analysis
Genomic DNA was extracted by standard methods using the Wizard Genomic DNA Purification System (Promega, Madison, WI). Bisulfite modification of genomic DNA (500 ng) was performed using the EZ DNA methylation kit (Zymo Research, Orange, CA), as per manufacturer’s instructions. The methylation level of APC and GSTP1 was assayed by PSQ. PCR and sequencing primers were designed using the PyroMark Assay design software (version 2.0.1.15; Qiagen, Valencia, CA). The PSQ assay was designed to evaluate the methylation status of four CpG sites in each gene. Primer sequences, amplification conditions, and untranslated DNA sequence information are described in
Statistical Analysis
The APC and GSTP1 methylation level of each sample was expressed as a mean value (the sum of each CpG site methylation level [%]/total number of CpG sites [n = 4]). To incorporate the overall extent of methylation, each patient’s methylation score (M score) was calculated as the sum of the methylation levels of the two genes multiplied by the corresponding odds ratios, derived from the logistic regression analysis assessing the predictive value of each gene for BPH and PCa. The quantitative methylation levels of APC and GSTP1 and the M score were compared to clinicopathological parameters. Differences in continuous variables between groups were assessed by two-sample t-test. Categorical variables were compared by a χ2 test. The optimal sensitivity and specificity of the APC methylation level, GSTP1 methylation level, and M score for the diagnosis of PCa were determined by receiver operator characteristic (ROC) curve analysis using MedCalc Software 12.0 (Mariakerke, Belgium). Using the same thresholds, sensitivity, specificity, positive predictive value, and negative predictive value were also calculated. Pearson’s correlation was used to evaluate the relationship between methylation level and clinicopathological parameters. Tests for trend were performed by analysis of variance (ANOVA) trend analyses using polynomial contrasts. For statistical purposes, PCa samples were divided into subgroups according to clinicopathological parameters as followed: (1) Gleason score (≤6, 7, ≥8), (2) stage at diagnosis (T2, T3, T4), and (3) PSA level at diagnosis (<10, 10–20, ≥20 ng/mL). Statistical analysis was performed using SPSS 12.0 software (SPSS, Inc., an IBM Company, Chicago, IL). p < 0.05 was considered statistically significant.
Results and Discussion
The baseline characteristics of all clinical specimens are presented in Table 1 . There was no significant difference in mean age or total prostate volume between the BPH and PCa patients. PCa patients had elevated levels of PSA compared with BPH patients. The methylation levels of APC and GSTP1 were significantly higher in the PCa samples compared with the BPH samples (31.9% ± 20.3% vs. 1.4% ± 2.1% and 50.0% ± 30.4% vs. 2.8% ± 3.1%, respectively, each p < 0.001; Fig. 1A , B ). To integrate the methylation status of APC and GSTP1, the M score was calculated for each patient. The M score was also significantly elevated in PCa patients compared with BPH patients (113.1 ± 60.9 vs. 5.7 ± 5.1, p < 0.001; Fig. 1C ). ROC analysis was conducted to assess the predictive value for PCa. The area under the curve reached 0.954, 0.942, and 0.983 for APC methylation, GSTP1 methylation, and M score, respectively ( Fig. 2 and Table 2 ). To ensure well-balanced sensitivity and specificity, the threshold of APC methylation, GSTP1 methylation, and M score was determined. Using that threshold, patients were divided into two groups: hypermethylated and unmethylated. As shown in Table 2 , the frequency of APC and GSTP1 methylation was 88.7% (86/97) and 91.8% (89/97) in PCa and 3.8% (2/52) and 3.8% (2/52) in BPH, respectively. When the M score was applied, no methylation was found in BPH samples: The M score methylation percentage in PCa and BPH was 92.8% (90/97) and 0% (0/52), respectively. After stratification by PSA level, stage, and Gleason score, the methylation frequency of APC, GSTP1, and M score showed similar results (data not shown).
Baseline Characteristics of Patients Included in the Study
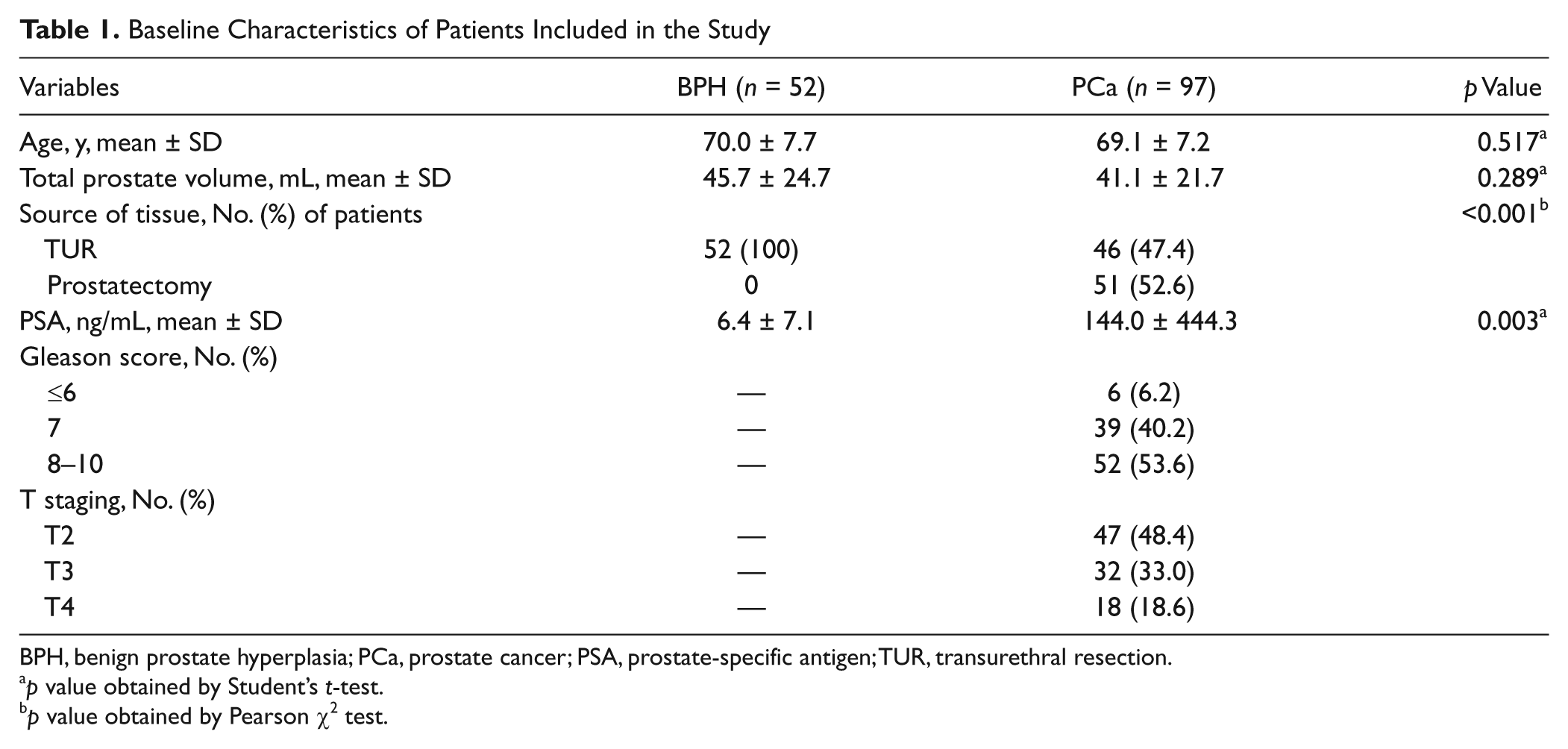
BPH, benign prostate hyperplasia; PCa, prostate cancer; PSA, prostate-specific antigen; TUR, transurethral resection.
p value obtained by Student’s t-test.
p value obtained by Pearson χ2 test.
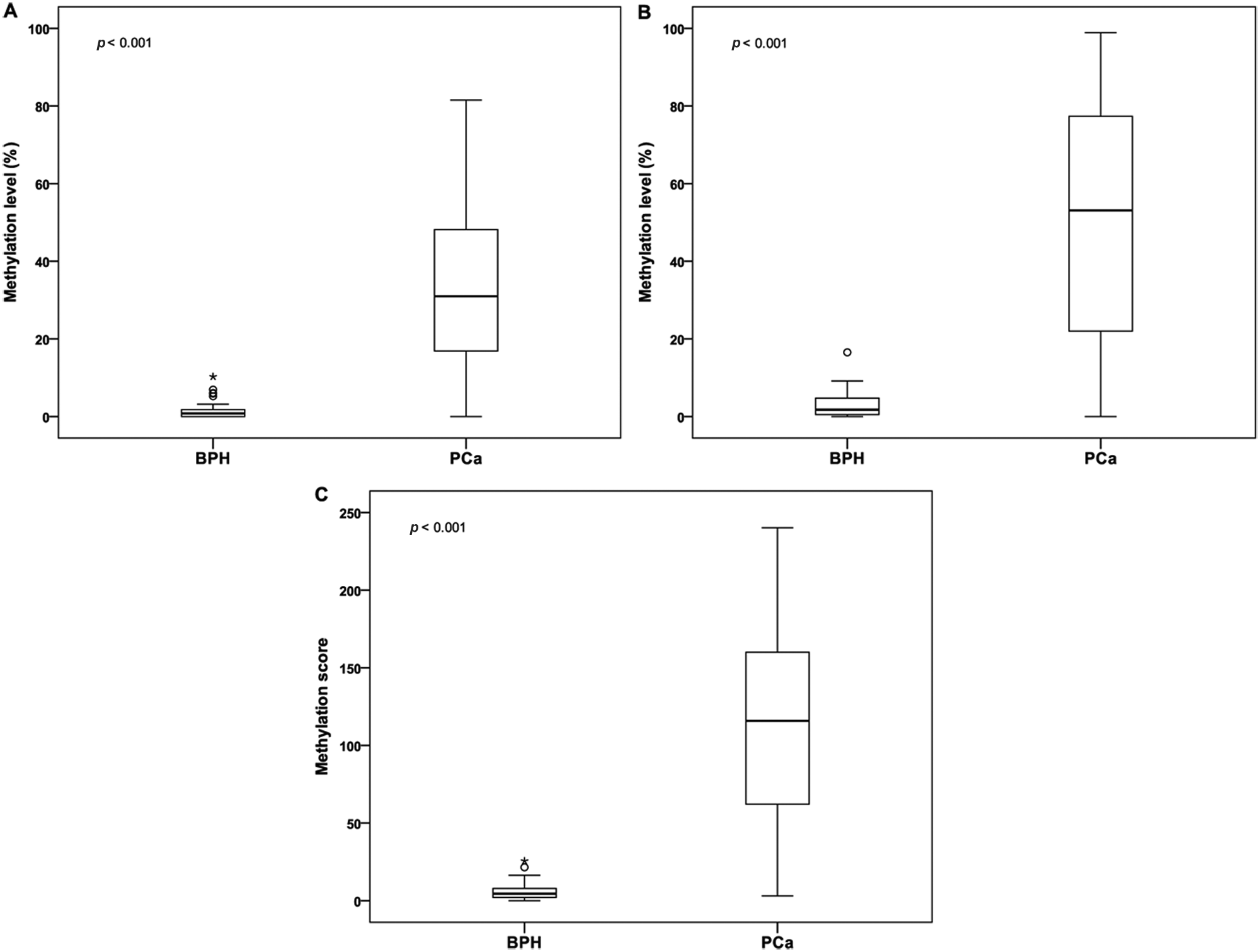
Methylation level of (
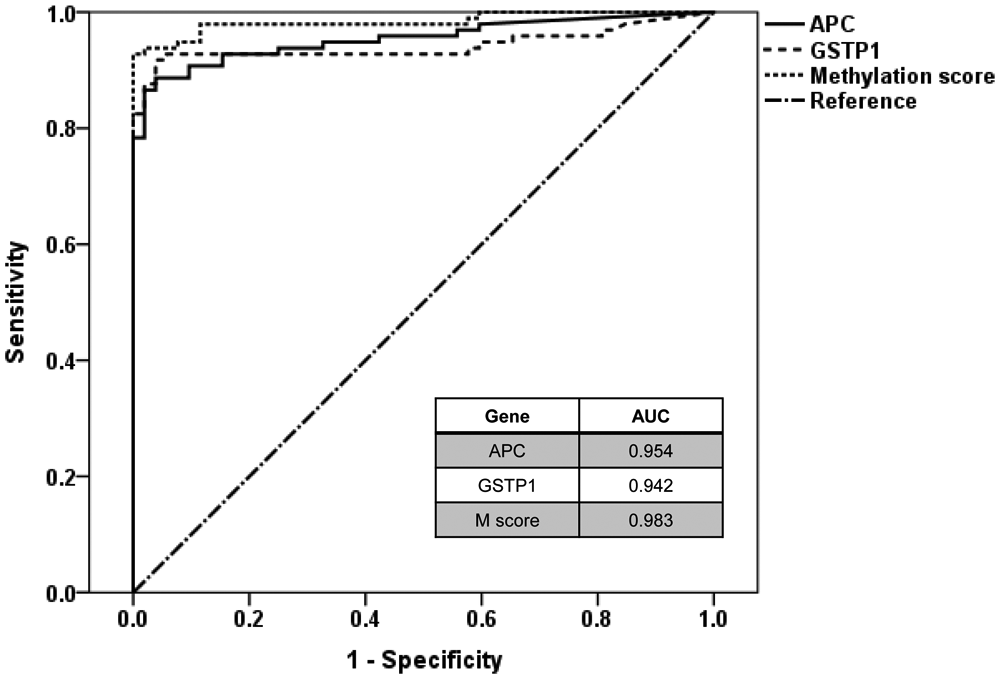
Receiver operator characteristic curve analysis of methylation markers for the prediction of prostate cancer. APC, adenomatous polyposis coli; GSTP1, glutathione-S-transferase-P1.
Frequency of Single and Combination Gene Methylation in Human Prostate Tissues
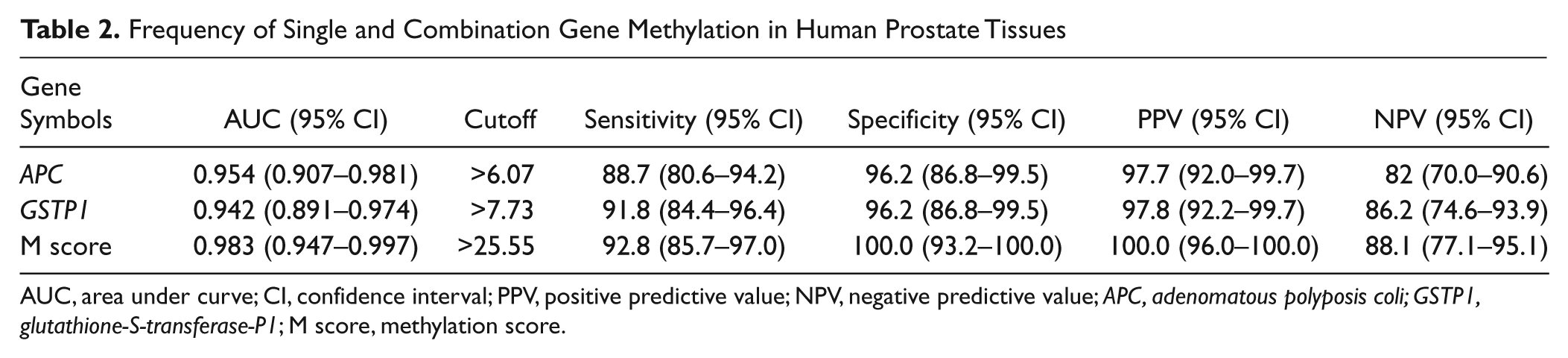
AUC, area under curve; CI, confidence interval; PPV, positive predictive value; NPV, negative predictive value; APC, adenomatous polyposis coli; GSTP1, glutathione-S-transferase-P1; M score, methylation score.
To evaluate the relationship between the methylation level of APC, the methylation level of GSTP1, the M score, and clinicopathological parameters, a correlation analysis was performed. No correlation was found between methylation and age, prostate volume, or Gleason score (data not shown). Preoperative serum PSA level showed a positive correlation with GSTP1 methylation level (r = 0.230, p = 0.005) and M score (r = 0.162, p = 0.049) but no correlation with APC methylation level. When subjects were divided according to Gleason score, tumor stage, and PSA level, the APC methylation level, GSTP1 methylation level, and M score were significantly associated with serum PSA level (p trend <0.001;
Abnormal DNA methylation patterns are associated with many human tumor types. Differences in methylation patterns have emerged as markers for cancer risk assessment, cancer diagnosis, and therapy monitoring in several different types of cancer.3,4 Moreover, DNA methylation for cancer detection is quite powerful due to the inherent stability of DNA compared with RNA or protein. 3 Recently, several methodologies have become available to detect the methylation status of certain genes in clinical samples,3,4 but the reported methylation levels are inconsistent, even between studies of the same gene in PCa specimens. Several factors may explain these discrepancies, including differences in sample types, analytical methods, or PCR methods.3,4 Accordingly, the focus has now shifted to those aberrant methylation events that are absent in normal cells and to the development of techniques that provide reliable, sensitive, and fast results to study these potential biomarkers.2–4 The use of conventional methylation-specific polymerase chain reaction (MSP) is limited in cancer detection because benign lesions can be weakly positive and cannot be distinguished from cancer cases. Moreover, the results of MSP in any particular DNA region are reported simply, and perhaps subjectively, as methylated or unmethylated, without allowing the quantitation or identification of partial methylation. The quantitative analysis of DNA methylation with appropriate methods might improve the accuracy of data interpretation obtained from small amounts of DNA in clinical samples. In that respect, PSQ might be a superior method because it provides quantitative, rather than qualitative, information for each target CpG site. In addition, the assay design allows the interrogation of different parts of a gene as well as the inclusion of internal controls for incomplete bisulfite conversion.4,5 To date, aberrant hypermethylation of CpG island-containing promoters has been identified at numerous loci in PCa, including GSTP1 and APC.7–11 However, relatively limited studies have used the quantitative PSQ method to explore DNA hypermethylation status of various genes in PCa.5,6 In the earlier studies using multiple genes by the PSQ method, it was demonstrated that both APC and GSTP1 methylation levels were significantly higher in PCa samples than in control samples.5,6 Our study confirmed the study method and results of previous quantitative analysis.5,6
Regarding study method and results, our study may be similar to those of earlier ones but introduced clear differences from previous studies.5,6 Previous studies evaluated methylation level of multiple genes individually and did not try to merge the methylation level of multiple genes in a quantitative nature, whereas we quantitatively integrated methylation levels of APC and GSTP1 to enhance its diagnostic and prognostic power as the biomarkers for PCa. The quantitative integration of the methylation levels of multiple genes is one of the major advantages of PSQ. Combinations allow the quantitative comparison of samples and the accurate segregation of pathologic covariates based on methylation levels. Merging the methylation levels of several genes yields maximal specificity and sensitivity. For the prediction of biologic behavior of tumor, the use of a single gene locus has several drawbacks. 11 First, the maximum sensitivity can only be as high as the frequency of hypermethylation at a specific CpG locus. Second, noncancerous tissues can in some cases harbor CpG island hypermethylation at the same gene locus. Third, the methylation of a single gene locus may occur in other cancers and thus be misleading in PCa. Furthermore, the number of hypermethylated genes has been shown to increase as PCa progresses; thus, cancer development, progression, and aggressiveness are likely to be best captured by a combination of markers. Previous studies have indeed demonstrated that using markers in combination provided greater diagnostic power than using a single marker.4,11,12 Similar to our study, Enokida et al. 12 first employed the M score to integrate the methylation status of multigenes, which is the sum of the log hazard ratios (HRs) analyzed by multivariate logistic regression analysis for the predictive value of each gene for BPH and PCa. In contrast to our quantitative study method, after analysis of electrophoresis in agarose gels, relative methylation levels (%) were calculated with ImageJ software (National Institutes of Health, Bethesda, MD) by using the area under the curve corresponding to the methylated or unmethylated PCR products band. In the present study, we found that the combination of the methylation level of these two genes maximizes the specificity and increases the sensitivity for discriminating between PCa and BPH.
APC and GSTP1 are well-characterized tumor suppressor genes. APC downregulates Wingless-type (Wnt) signaling by targeting the transcriptional coactivator b-catenin for proteasomal degradation, thereby preventing its association with the nuclear transcription factor TCF/LEF. 13 The Wnt pathway plays a central role in tumorigenesis. Its inappropriate activation is a common feature of many human cancers, leading to the deregulation of cell proliferation and differentiation. 14 GSTP1 is involved in the metabolism, detoxification, and elimination of potentially genotoxic foreign compounds and thus acts to protect cells from DNA damage and cancer initiation. Suppression of GSTP1 activity can result in enhanced susceptibility to DNA damage and increased cancer incidence. 10 The close association of APC and GSTP1, either alone or together, with PCa has been reported.7,8,10,11 Although it is typically relatively high in PCa and low in BPH, the reported strength of the association varies widely between studies.7,8,10,11 As previously mentioned, the inconsistency in methylation results for these genes may be due to differences in detection methods, regions of interest, and/or tissue types. In the present study, the combination of APC and GSTP1 methylation by a quantitative method yielded a sensitivity of 92.8% while maintaining a specificity of 100% for the discrimination of PCa samples from benign prostate samples. This finding underlies the importance of appropriate detection methods and gene combinations. Our findings are consistent with previous results and demonstrate that APC and GSTP1 hypermethylation is a reliable predictive indicator of PCa.7,8,10,11
Although histologically confirmed prostate tissues were used, the possibility of unrevealed PCa in BPH patients and that of undetected small fractions of methylated DNA molecules might affect the sensitivity and specificity of our results. Nonetheless, promising methylation frequency results were obtained. Although these findings are promising, multicenter large-scale clinical validation studies using primary human cancer tissues and body fluids are currently under way at our institute to confirm this two-gene methylation signature as a suitable diagnostic methylation marker for PCa. These studies will improve our understanding of the biological role and clinical relevance of methylation in tumorigenesis.
Conclusively, our study demonstrates that the combination of methylation markers with a quantitative detection method might drastically improve the ability to discriminate PCa from BPH. In particular, the combination of APC and GSTP1 methylation levels may be a useful marker of PCa.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2011-0003333 and 2011-0001044); and a grant from the Next-Generation BioGreen 21 Program (No. PJ0081952011), Rural Development Administration, Republic of Korea.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.