
Abstract
In recent years, HIV-1 integrase (IN) has become an established target in the field of antiretroviral drug discovery. However, its sole clinically approved inhibitor, the integrase strand transfer inhibitor (INSTI) raltegravir, has a surprisingly low genetic barrier for resistance. Furthermore, the only two other integrase inhibitors currently in advanced clinical trials, elvitegravir and dolutegravir, share its mechanism of action and certain resistance pathways. To maintain a range of treatment options, drug discovery efforts are now turning toward allosteric IN inhibitors, which should be devoid of cross-resistance with INSTIs. As IN requires a precise and dynamic equilibrium between several oligomeric species for its activities, the modulation of this equilibrium presents an interesting allosteric target. We report on the development, characterization, and validation of an AlphaScreen-based assay for high-throughput screening for modulators of HIV-1 IN dimerization. Compounds identified as hits in this assay proved to act as allosteric IN inhibitors. Additionally, the assay offers a flexible platform to study IN dimerization.
Introduction
Integration of retroviruses such as HIV-1, the causative agent of AIDS, establishes a provirus in the human genome and constitutes the point of no return in viral replication. It is a multistep process catalyzed by the viral integrase (IN) in cooperation with cellular cofactors and completed by the cellular DNA repair machinery. 1 IN is a 32 000–Da enzyme encoded by the viral pol gene. It consists of three canonical domains that are found in all retroviral integrases: the N-terminal, catalytic core, and C-terminal domains (NTD, CCD, and CTD, respectively).2,3
During the integration process, IN catalyzes two major reactions that are spatially and temporally separated. 1 In the first reaction, termed 3′ processing (3P), a dinucleotide is cleaved from the 3′ ends of the viral genome. 1 This exposes a conserved 3′ CA dinucleotide critical for the second reaction: strand transfer (ST), which can only occur after import of the viral preintegration complex into the nucleus. 4 In this reaction, IN concertedly inserts both viral 3′ ends into opposing strands of the target DNA major groove. 2 Depending on the retrovirus, a typical stagger of four to six nucleotides remains. The resulting two single-stranded gaps are repaired by host cell machinery, establishing a stably integrated provirus. 2 IN does not simply function as a monomer in these reactions; in solution, it is present in an equilibrium of monomers, dimers, tetramers, and even higher multimeric species. To complicate matters even further, these species are unequal in their catalytic activities: 3P requires at least a dimer, whereas ST activity is only evident for the IN tetramer.1,5
Around 30 years of research have spawned 26 U.S. Food and Drug Administration–approved molecules to combat AIDS. As large as this repertoire might appear, rapid evolution aids HIV to quickly overcome the genetic barriers associated with antiviral resistance. The current standard of care for patients, highly active antiretroviral therapy (HAART), hence entails taking combinations of at least three antiretrovirals with at least two different mechanisms of action to prevent resistance. The error-prone nature of reverse transcription and HIV’s vast replication potential force us to investigate new targets for the development of inhibitors. Of the current antiretrovirals, only one targets IN: raltegravir (MK-0518), which was approved for use in treatment-experienced patients in 2007 and in treatment-naive patients in 2009. 6 Two other integrase strand transfer inhibitors (INSTIs) are currently in phase III clinical trials: elvitegravir (GS-9137) and dolutegravir (GSK-572).7,8 Importantly, all of these molecules have a common mechanism of action: they bind to the complex of IN with 3′ processed viral DNA, displacing the catalytic adenosine and, as such, disarming the complex and blocking ST. 2 As a consequence, significant cross-resistance is observed, even between first-generation (raltegravir, elvitegravir) and second-generation compounds (e.g., dolutegravir). 9
These findings have encouraged IN-targeted drug discovery to move away from the active site and focus on allosteric inhibitors instead.10,11 A recent feat in this respect was the discovery and development of LEDGINs, a class of small molecules inhibiting the interaction of IN with its cellular cofactor, lens epithelium-derived growth factor (LEDGF/p75). 12 LEDGINs bind to a site distinct from the catalytic site and show no cross-resistance to any known IN inhibitors. As IN requires a dynamic equilibrium between at least dimers and tetramers for its activities, another candidate target for the design of allosteric IN inhibitors is the oligomerization of the enzyme. 13 Two different approaches have already provided proof-of-principle results. First, peptides derived from the IN CCD dimer interface were described to effectively compete with IN dimer formation.14,15 Some of these peptides (INH1 and INH5, referring to α-helices 1 and 5 of the CCD) were able to shift the IN oligomerization equilibrium entirely to the monomer. 14 Second, molecules have been identified that bind across the IN dimer interface, establishing contacts with both monomers.16–19 Consequently, these compounds stimulate dimerization and shift the oligomerization equilibrium to certain species, resulting in a loss of catalytic activity.13,16,20–22
Recently, a homogeneous time-resolved fluorescence-based (HTRF) assay measuring IN dimerization was reported and used in combination with mathematical modeling to study the dynamics of IN-IN and IN-LEDGF/p75 interactions.17,18 This assay, however, to our knowledge, has not been validated for high-throughput screening (HTS) or used for the discovery of small-molecule inhibitors of IN. Hence, aforementioned studies and the inclination toward allosteric IN inhibitors encouraged us to develop an AlphaScreen-based IN dimerization assay suited for these purposes (outlined in Fig. 1 ). This assay detects all dimerization modulators, both inhibitors and stimulators, and can also be used to study IN dimerization itself. Last, to provide “cleaner” hit lists during drug discovery, we implemented a counterscreen to eliminate molecules that interfere with the AlphaScreen technology itself.
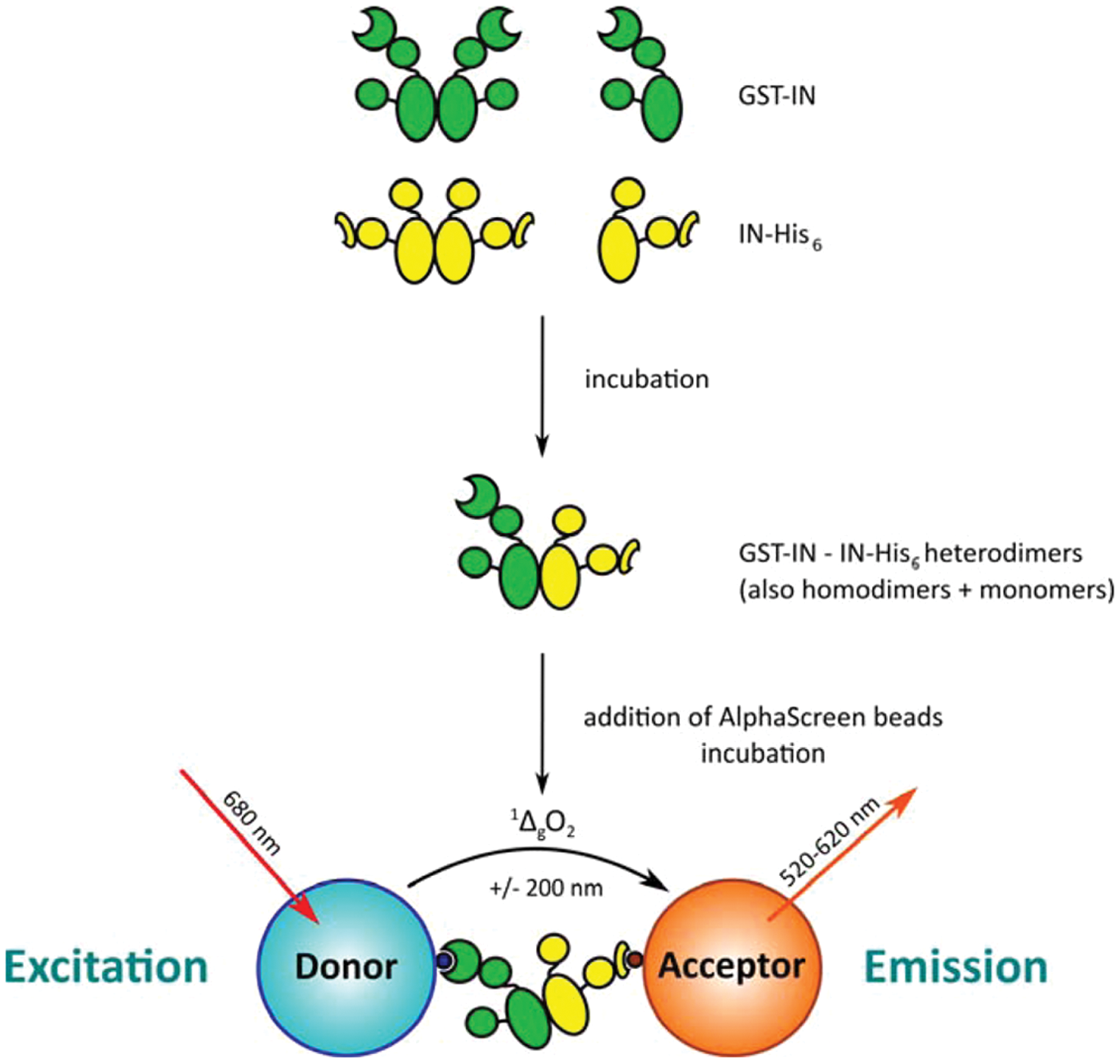
Overview of the integrase (IN) dimerization AlphaScreen assay. Glutathione S-transferase (GST)–tagged IN (green) is mixed with His6-tagged IN (yellow) at the desired concentrations. Incubation at 4 °C allows monomer exchange between dimers and the establishment of a steady-state population containing GST- and His6-homodimers as well as GST/His6-heterodimers. When Ni2+-chelate acceptor and glutathione donor AlphaScreen beads are added, the heterodimers will bring both beads into close proximity. Upon irradiation with 680 nm laser light, a photosensitizing phthalocyanine in the donor beads will produce large amounts of singlet oxygen (1ΔgO2). Due to the short half-life of this species (±4 µs), it can only diffuse about 200 nm before returning to the ground state. If, however, acceptor beads are nearby because of interactions of the biomolecules on their surface, the 1ΔgO2 will reach these beads and react with thioxene-based groups on the inside. Energy transfer to anthracene-based and then rubrene-based groups will ultimately result in emission of light between 520 and 620 nm: the AlphaScreen signal. Changes in the population of heterodimers (e.g., due to the presence of modulating compounds) will, upon addition and incubation with the beads, result in an altered output signal and can be picked up.
Materials and Methods
Construction of Protein Expression Plasmids
The full-length HIV-1 IN coding sequence was amplified by PCR from pINSD.His by using forward and reverse primers encoding BamHI and XhoI recognition sites, respectively (F: 5′-TTGGATCCTTTTTAGATGGAATAGATAAGGC-3′ and R: 5′-AAAACTCGAGCTAATCCTCATCCTGTC-3′). The cleaved fragment was then ligated into the corresponding sites of pGEX-6P-1, creating the pGEX-6P-1-IN construct for expression of recombinant glutathione S-transferase (GST)–tagged IN protein.
The pGEX-6P-2-His plasmid for expression of recombinant His6-tagged GST (GST-His6) protein used in the AlphaScreen counterscreen was constructed through adapter ligation. Oligos (5′-GATCGCATCACCATCACCATCACCATCACCATTCCCC-3′ and 5′-GATCGGGGAATGGTGATGGTGATGGTGATGGTGATGC-3′) were dissolved in annealing buffer (10 mM Tris [pH 7.5], 50 mM NaCl, 1 mM EDTA), mixed, and heated to 95 °C for 5 min. The mixture was then left to cool slowly to room temperature (RT) in the heating block. The annealed adapter was ligated into the BamHI site of pGEX-6P-2, and its correct orientation was verified by sequencing.
Recombinant Protein Purification
GST-tagged wild-type (WT) or A128T HIV-1 IN was purified as follows. A preculture was prepared by inoculating 50 mL Lysogeny broth (LB) medium containing 100 mg/L ampicillin and 25 mg/L chloramphenicol with BL21 CodonPlus-competent cells harboring the pGEX-6P-2-IN or pGEX-6P-2-INA128T plasmid. This culture was left to shake at 37 °C overnight (ON). The next day, it was transferred into 2.5 L fresh medium and incubated until the OD reached 0.6. Protein production was induced by addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and the temperature was lowered to 30 °C. After 3 h, the culture was harvested by spinning down for 10 min at 5000 rpm. Cells were washed in 20 mL STE buffer (10 mM Tris [pH 7.5], 100 mM NaCl, 0.1 mM EDTA), spun down again, and the pellet stored at −20 °C. The cells were lysed by 6 × 1–min sonication in 20 mL buffer A (25 mM HEPES [pH 7.5], 500 mM NaCl, 1 mM MgCl2, 7.5 mM 3-[(3- cholamidopropyl)dimethylammonio]-1-propanesulfonate [CHAPS], and 5 mM dithiothreitol [DTT]), supplemented with 1 mM phenylmethanesulfonyl fluoride (PMSF), 1 U/10 mL DNase, and an additional 7.5 mM CHAPS. The lysate was cleared by 30 min of centrifugation at 15 000 rpm and 4 °C, after which the supernatant was applied onto 2 mL of previously washed (10× bed volume, buffer A) Glutathione Sepharose 4 Fast Flow (GE Healthcare, Fairfield, CT). After the cleared lysate had flown through, the column was washed with buffer A (10× bed volume), and protein was eluted with 10 mL buffer A supplemented with 25 mM GSH. Peak fractions were pooled and dialyzed against 100× excess of buffer A containing 10% glycerol. Aliquots were made and flash-frozen in liquid nitrogen.
C-terminally His6-tagged WT or A128T HIV-1 IN, encoded by pET-20b-IN or pET-20b-INA128T, respectively (pKBIN6H or pKBIN6HA128T), was purified by affinity and ion exchange chromatography as previously described. 19
The double-tagged GST-His6 protein was produced using standard conditions. Briefly, a 1 L culture of BL21 CodonPlus cells harboring the pGEX-6P-2-His plasmid was grown to OD 0.6 and induced for 3 h at 37 °C with 1 mM IPTG. Bacteria were then harvested, washed with STE buffer as described above, and the pellet stored at −20 °C. Cells were lysed by sonication for 6 × 1 min in 10 mL buffer B (standard phosphate-buffered saline, 5 mM DTT, 40 mM imidazole) supplemented with 1 mM PMSF and 1 U/10 mL DNase. The lysate was cleared through 30 min of centrifugation at 15 000 rpm and 4 °C, and the supernatant was applied onto 2 mL washed (10× bed volume, buffer B) His-Select Nickel affinity gel (Sigma-Aldrich, St. Louis, MO). After another washing step (10× bed volume, buffer B), protein was eluted with 10 mL buffer B containing 250 mM imidazole. Again, peak fractions were pooled and dialyzed against 100× excess of buffer B with 10% glycerol. Aliquots were made and flash-frozen in liquid nitrogen.
For all recombinant proteins, purity was checked by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Concentrations were estimated by measuring UV absorption (A280) and applying the Beer-Lambert law with calculated molar extinction coefficients (ϵ280).
IN Dimerization Assay
The IN dimerization assay is based on the robust AlphaScreen technology platform (Amplified Luminescent Proximity Homogeneous Assay, ALPHA; PerkinElmer, Waltham, MA), which derives from the Luminiscent Oxygen Chanelling Immunoassay (LOCI) originally described by Ullman et al. 23 Briefly, the technology works as follows. Key to the assay are two types of ±250-nm diameter, chemical-containing, hydrogel-coated latex beads: the AlphaScreen donor and acceptor beads. Each can be coated with a protein of interest. When irradiated with 680 nm laser light, a photosensitizing phthalocyanine in the donor beads generates large amounts of singlet oxygen (1ΔgO2). Due to its short half-life (±4 µs), 1ΔgO2 only diffuses about 200 nm before returning to the ground state. If, however, an acceptor bead is nearby because of interaction of the biomolecules on its surface with those on the surface of the donor bead, the 1ΔgO2 will permeate these beads and react with thioxene-based groups on the inside. Energy transfer to anthracene-based and then rubrene-based groups ultimately results in emission of light between 520 and 620 nm from the acceptor beads, which is detected as the AlphaScreen signal and measured in photon counts per second.
Our assay was optimized for use in 384-well OptiPlate microplates (PerkinElmer) with a final volume of 25 µL. Compounds and proteins were all diluted to 5× working solutions in the assay buffer (25 mM Tris [pH 7.4], 150 mM NaCl, 1 mM MgCl2, 1 mM DTT, 0.1% [v/v] Tween-20, and 0.1% [w/v] bovine serum albumin [BSA]). First, 5 µL of the inhibitor, buffer, or control was pipetted into the wells, followed by 5 µL GST-IN and 5 µL IN-His6 working solutions. The plate was sealed and left to incubate for 3 h at 4 °C. This step allows an equilibrium situation to be established while the low temperature counteracts IN aggregation. Next, 10 µL of a mix of Ni2+-chelate acceptor and glutathione donor AlphaScreen beads (PerkinElmer) was added. This brings the total volume to 25 µL and establishes final concentrations of 10 µg/mL for each of the beads and 15 nM for each IN protein. Compounds are usually assayed at 100 µM final, resulting in 0.4% DMSO, which is well within the tolerance levels of the assay (data not shown). After addition of the beads, the plate was placed at RT and incubated for 2 more h before being read in the EnVision Multilabel Reader (PerkinElmer) in AlphaScreen mode (for an overview of the assay, see Fig. 1 ). Assay quality is controlled through the use of several controls: one negative control (buffer containing 0.4% DMSO) and two positives (100 µM INH5 peptide, obtained from Pepscan [Lelystad, the Netherlands], as inhibitor 14 and 100 µM LEDGIN 7, synthesized as described by Christ et al., 12 as stimulator). Data were analyzed with Prism 5.0 (GraphPad Software, La Jolla, CA) and/or Excel (Microsoft, Redmond, WA).
IN Dimerization Assay Cross-Titration
The assay was performed as described above with some modifications: both proteins were titrated against each other (1000, 300, 100, 30, 10, 3, 1, and 0 nM), the first incubation step was performed ON at 4 °C, and a final concentration of 20 µg/mL beads was used.
IN Dimerization Assay Beads Optimization
Optimization of the amount of AlphaScreen beads to reduce cost per well while acknowledging the signal-to-background (S/B) ratio was performed through small modification of the general protocol described above. Dilution series of both proteins (in a 1:1 ratio) were incubated ON at 4 °C, after which different amounts of AlphaScreen beads were added (20, 10, 5, 2.5, or 0 µg/mL final). After reading the plate, the S/B ratio was calculated by taking the average maximal signal (100 nM protein) and dividing by the average background signal (0 nM protein) for that bead concentration.
IN Dimerization Assay Time Course
Time course experiments were performed as follows: at time 0, GST-IN and IN-His6 proteins were mixed at the desired concentrations in assay buffer (133.3, 66.7, 33.3, 16.7, and 0 nM in order to give 80, 40, 20, 10, and 0 nM final). The mixtures were kept slowly shaking at 4 °C. At the indicated time points (15, 30, 60, 120, 240, 300, 360, and 420 min), samples were taken and 15-µL aliquots were pipetted into wells of the 384-well plate. Again, 10 µL of a mix of Ni2+-chelate acceptor and glutathione donor AlphaScreen beads was added, bringing the total volume per well to 25 µL, beads to 10 µg/mL, and proteins to the concentrations mentioned above. For each time point and concentration, three replicates were prepared. The plate was covered, shielded from light, and set to incubate for 2 h at RT. Finally, the plate was read on the EnVision Multilabel Reader. This allowed us to estimate the minimal time required for full equilibration of the IN population present in the samples at around 180 min.
GST-His6 AlphaScreen Counterscreen
All components (GST-His6 protein, controls, compounds, and beads) were diluted to their respective working dilutions in the assay buffer described above. Then, 5 µL of the compound under study was pipetted into a well of a 384-well OptiPlate, followed by 10 µL GST-His6 and 10 µL of a Ni2+-chelate acceptor glutathione donor beads mixture. The final volume was 25 µL and contained 10 nM GST-His6, 100 µM of the compound under study, and 10 µg/mL of each bead. The plate was shielded from light, incubated for 1 h at RT, and read on the EnVision Multilabel Reader. Any compound emerging as a hit in this assay likely interferes with the AlphaScreen technology itself, either by absorption of excitation or emission light, singlet oxygen quenching, chelation of Ni2+, or other mechanisms. Quality of the screen is monitored and results are normalized through two controls: a negative control (0.4% DMSO) and a positive one (100 µM bromophenol blue, which absorbs acceptor beads emission light of 520-620 nm).
Overall IN Catalytic Activity ELISA
The enzymatic integration reactions were carried out with minor modifications as described before.12,24 To determine the susceptibility of HIV-1 IN to different compounds, we used an enzyme-linked immunosorbent assay (ELISA). The overall integration assay uses an annealed oligonucleotide substrate, one strand of which (5′-ACTGCTAGA GATTTTCCACACTGACTAAAAGGGTC-3′) is labeled with biotin at the 3′ end and the other (5′-GACCCTTTTAGTCAGTGTGGAAAATCTCTAGCAGT-3′) is labeled with digoxigenin at the 5′ end. IN was diluted to 1.6 µM in 10 mM Tris (pH 7.6), 750 mM NaCl, 10% (v/v) glycerol, and 1 mM 2-mercaptoethanol. To perform the reaction, 4 µL of diluted IN and the desired amount of compound were added to a final volume of 36 µL containing 20 mM HEPES (pH 7.5), 10 mM MgCl2, 5 mM DTT, 5% (v/v) polyethylene glycol, and 15% (v/v) DMSO. After a 30-min incubation, 4 µL annealed oligonucleotide (7 nM) was added, bringing the total volume to 40 µL and the concentration of IN to 160 nM. The reaction was carried out for 1 h at 37 °C. Reaction products were denatured with 30 mM NaOH and detected by ELISA on avidin-coated plates.
Results
We here describe the development of an assay to screen for small-molecule modulators of the HIV-1 IN oligomerization equilibrium. Our assay is based on the AlphaScreen technology platform as it combines sensitivity and adaptability with a very low background, even when working with an enzyme that is prone to aggregate such as HIV-1 IN. 5 The high S/B ratios allow for miniaturization and reagent stretching, ultimately leading to a cost-efficient assay. Another advantage of AlphaScreen is its versatility, enabling us to test different combinations of protein tags for the dimerization assay. While selecting a pair of tags for IN, we considered factors such as ease, yield and purity of purification, the availability of standard AlphaScreen beads for binding of the tags, and the final S/B ratio of the assay. Ultimately, we decided on an N-terminal GST-tag on one IN protein preparation together with a C-terminal His6-tag on the other.
Development of an IN Dimerization Assay
When both IN preparations are mixed in the assay, initially no or a very low AlphaScreen signal is expected as only GST/GST and His6/His6 IN dimers are present, which are unable to bring donor and acceptor beads together (see Fig. 1 ). The first incubation step of the mixture is critical in that it allows time for monomer exchange between these dimers and thus formation of the signal-inducing, double-tagged heterodimers. An estimated half-life of ±87 min for the IN dimer was determined by Tsiang et al. 17 in their HTRF-based assay. However, we could not simply extrapolate this value to our setup, as conditions are far from identical. As a consequence, initial tag optimization and other experiments performed before the time course study included an ON incubation step to ensure full equilibration of the system.
Development of most assays begins with the identification of an appropriate assay buffer. Previous experience with the study of HIV-1 IN interactions in an AlphaScreen format (e.g., with LEDGF/p75) allowed us to readily adapt buffers for the current assay. 12 Next, we carried out cross-titration experiments where both interacting proteins were titrated against each other. These experiments are performed to define optimal concentrations and ratios of proteins to be used in the final assay, to have a robust signal with as little protein as possible and staying well out of the “hooking” range. The hook effect occurs when the concentration of one of the interacting proteins is increased beyond saturation of its cognate bead. In such case, there will be free protein in solution that can bind to its partner on the other bead, effectively competing with protein-mediated bead-bead interactions and lowering the AlphaScreen signal accordingly. Results from a representative cross-titration are shown in Figure 2 . The effect of hooking can clearly be seen when concentrations reach 300 nM and higher, especially along the GST-IN axis. In the lower concentration range (<100 nM), the signal is optimal along the 1:1 (GST-IN:IN-His6) axis, justifying the use of this ratio for further experiments.
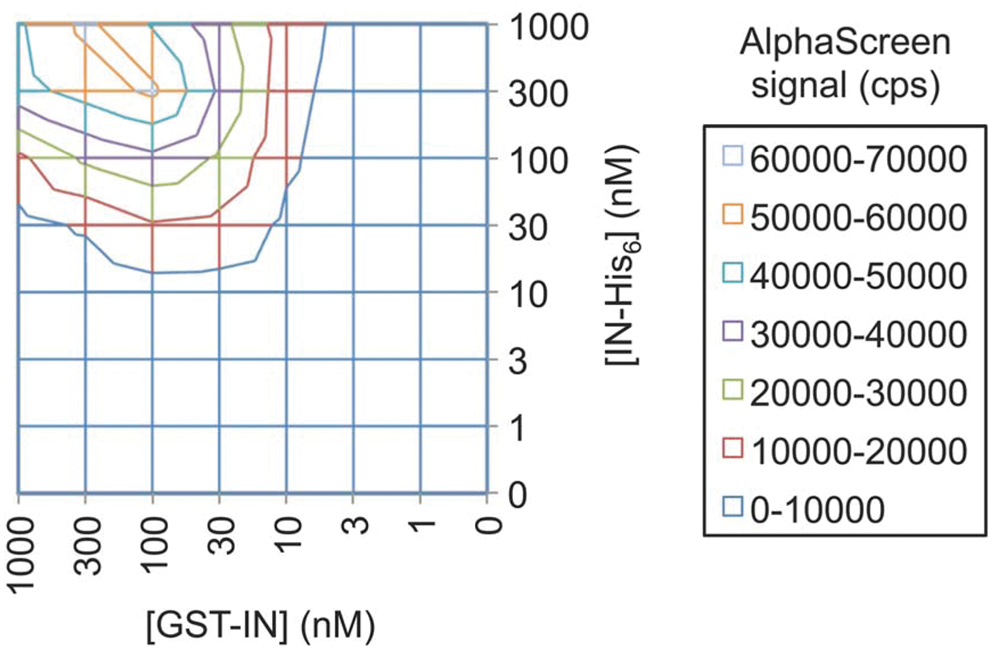
Cross-titration of GST-IN and IN-His6. GST-IN and IN-His6 are titrated against each other, starting from 1000 nM. The AlphaScreen signal arises most efficiently along the 1:1 axis in the lower concentration range (up to 100 nM), whereas hooking only appears at concentrations of 300 nM and higher. This is most evident with increasing concentrations of GST-IN. GST, glutathione S-transferase; IN, integrase.
The next optimization step aimed to reduce assay costs per well while still maintaining an assay that is robust enough to support HTS. Costs derive mainly from the amount of AlphaScreen beads used. Consequently, we studied the effect of lowering the amount of beads added on the S/B ratios in the assay. A dilution series of both proteins in a 1:1 ratio was incubated with different amounts of AlphaScreen beads and the signals were analyzed. Results of these dilution series are shown in Figure 3A . Lowering the amount of beads did not have an effect on the overall equilibrium of the assay, as the curves start to level off around the same concentration of protein. However, it significantly influenced the maximal (at 100 nM protein) and background signals (0 nM protein). As a result, within our tested range, the S/B ratio decreased linearly with the amount of beads ( Fig. 3B ). Considering this reduction in assay quality that accompanies the use of lower amounts of beads, we decided to decrease the amount of beads added only twofold, to 10 µg/mL. This way, costs are reduced almost twofold, but assay quality remains sufficiently high.
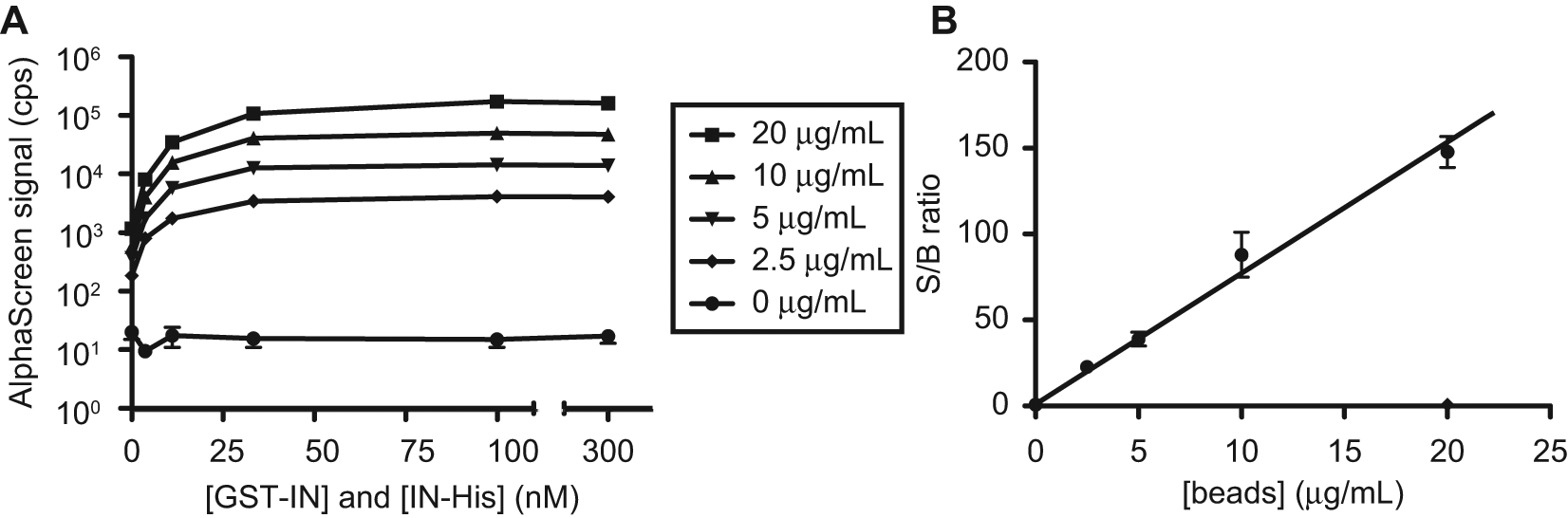
Optimization of the amount of AlphaScreen beads and signal-to-background (S/B) ratios. Dilution series of GST-IN and IN-His6 in a 1:1 ratio were incubated with different amounts of AlphaScreen beads and analyzed. (
IN Dimerization Assay Characterization
To characterize the assay further and investigate its kinetics, we performed time course experiments. In these studies, both IN proteins were mixed at various concentrations (1:1 ratio) and incubated at 4 °C. At certain time points, aliquots were transferred to the 384-well microtiter plate, beads were added, and the plate was incubated at RT. After 2 more hours the signal in these wells was measured. The data sets were fit with a one-phase exponential association model, and the resulting curves are shown in Figure 4 . Protein concentration did not significantly influence the observed association rate in the assay. We envision that binding of IN to the added AlphaScreen beads, as well as the avidity of the IN-IN mediated binding events between beads in particular, would significantly slow down progression toward the oligomerization equilibrium. This allowed us to derive an average apparent half-life for the exchange reaction of 22 ± 3 min. We also set a lower limit of 3 h for the initial assay incubation step, to achieve full equilibration of the system and hence a stable population of GST/His6-tagged heterodimers.
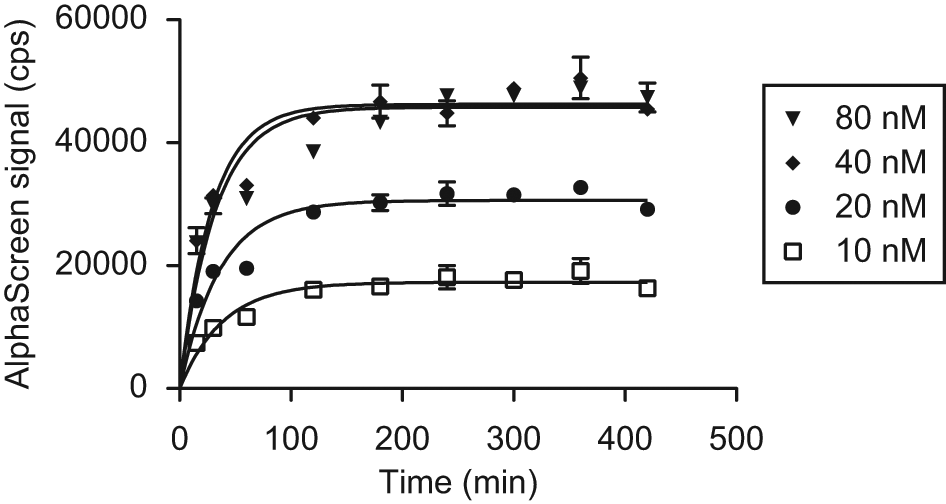
Time course of the IN dimerization assay. GST-IN and IN-His6 were mixed at t = 0 at different concentrations in a 1:1 ratio and incubated. Aliquots were transferred to a microtiter plate at the indicated time points, and beads were added. After another 2 h of incubation, the plate was read. The one-phase exponential association model used to fit the data shows that assay kinetics seems independent of the amount of protein added. The average half-life of the association reaction is estimated at 22 ± 3 min. We determined a minimal initial incubation time of 3 h to allow full equilibration of the system before AlphaScreen beads are added. GST, glutathione S-transferase; IN, integrase.
To evaluate the assay’s potential to discover inhibitors of IN dimerization, we tested the INH5 peptide described by Maroun et al. 14 This peptide, derived from the α5 helix of the HIV-1 IN CCD, was shown to bind the monomer-monomer interface and inhibit IN dimer formation. However, we also wanted to assess to which extent the optimized assay could identify compounds that bind the dimer across the monomer-monomer interface and hence stimulate dimerization. To test this hypothesis, we opted for LEDGIN 7, described by Christ et al. 12 LEDGIN compounds were designed as inhibitors of the LEDGF/p75-IN interaction but also display an additional allosteric inhibition of IN catalytic activity. This compound class has been thoroughly characterized, and its binding site is formed by both monomers constituting the IN dimer. Being an obligate dimer ligand, we investigated whether addition of LEDGIN 7 would affect IN dimerization in the assay. To control for unspecific effects, we also tested its effect on IN carrying the A128T LEDGIN resistance mutation (GST-INA128T and INA128T-His6). 12 In addition, we included the INSTI raltegravir, which should not bind efficiently to IN without processed DNA and hence should not show any effect in this assay. The compounds were first titrated, and the results, normalized to no-compound and no-protein controls (100% and 0% signal, respectively), are depicted in Figure 5 . For the antagonist INH5, we determined an EC50 of 4.5 µM (95% confidence interval [CI], 2.7–7.5 µM), whereas LEDGIN 7 behaved as an agonist in this assay, raising the signal to about 350% with an EC50 of 0.67 µM (95% CI, 0.35–1.29 µM). LEDGIN 7 was less potent in stimulating A128T IN dimerization, showing an EC50 of 2.6 µM (95% CI, 2.0–3.5 µM), or a fourfold increase over WT. As expected, raltegravir did not show any effect on dimerization. For comparison, LEDGIN 7 inhibited the IN-LEDGF/p75 interaction in AlphaScreen with an IC50 of 0.58 ± 0.3 µM and viral replication with EC50 and CC50 values of 0.76 ± 0.08 and 72.16 ± 5.15 µM, respectively. 12 The approximate fourfold increase in EC50 observed with A128T IN is in line with previously obtained in vitro results (F. Christ, unpublished results). The INH5 peptide was originally reported to show EC50 values of 85 and 60 nM for 3P and ST, respectively, against WT IN with Mg2+ and 11 and 4.7 µM against the more soluble F185K, C280S mutant with Mn2+ as a cofactor. 14 More recently, Tsiang et al. 17 reported an EC50 of 88 µM and calculated a Ki of 1.8 µM for this peptide in their HTRF-based assay.
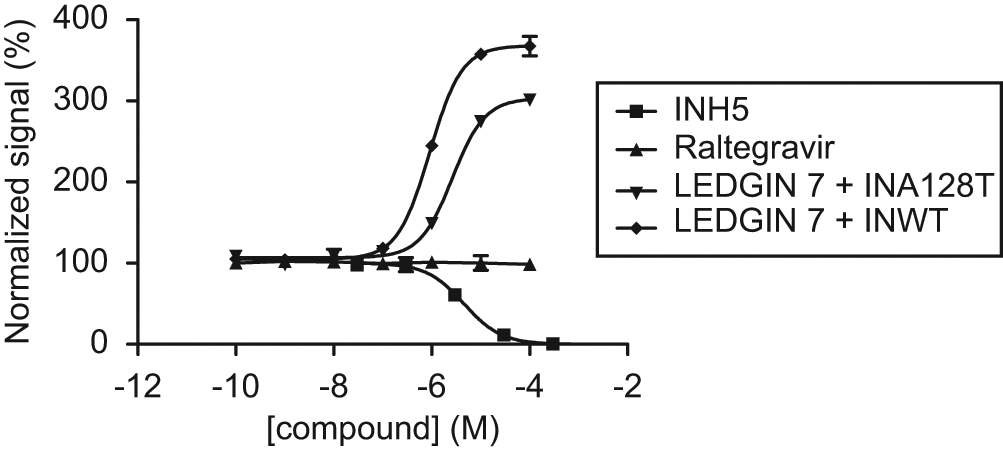
Titration of control compounds. The compounds INH5, LEDGIN 7, and raltegravir were titrated in the optimized IN dimerization assay against wild-type (WT) integrase (IN). Results were normalized by setting the no-compound and no-protein controls to 100% and 0%, respectively. Whereas INH5 acted as an antagonist with an EC50 of 4.5 µM, LEDGIN 7 behaved as an agonist in this assay with an EC50 of 0.67 µM. MK-0518 did not show any effect at all. When LEDGIN 7 was titrated against IN carrying the A128T resistance mutation, its EC50 was increased approximately fourfold to 2.6 µM, reflecting a reduced ability to stimulate IN dimerization.
Next, we prepared a test plate to ensure the suitability of our IN dimerization assay for HTS. The plate contained 32 wells for each of the three controls included (negative, 0.4% DMSO; stimulation, 100 µM LEDGIN 7; inhibition, 100 µM INH5). Results are shown in Figure 6 . We calculated Z′ factors 25 (Z′) and strictly standardized mean differences 26 (SSMD, β) for both stimulators and inhibitors. The obtained Z′ factors of 0.59 and 0.77, as well as SSMDs of 9.1 and −16.2, all indicate the presence of excellent windows for detection of both stimulators and inhibitors, respectively.
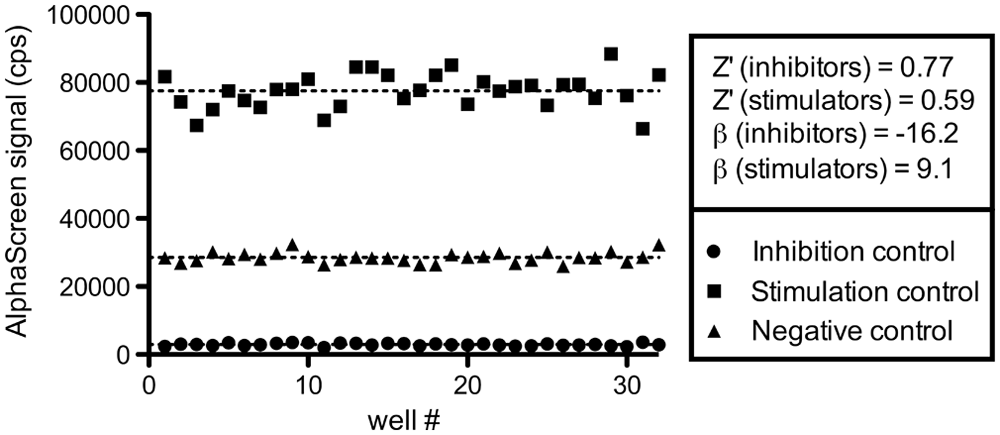
Integrase (IN) dimerization assay quality control. We evaluated the performance of our assay by preparing a test plate containing 32 wells of each of the following controls: an inhibition control (100 µM INH5), a stimulation control (100 µM LEDGIN 7), and a negative control (0.4% DMSO). Z′ factors were calculated to be 0.77 and 0.59 for the inhibition and stimulation window, respectively. 25 We also determined strictly standardized mean differences (SSMD, β) for both controls of −16.2 and 9.1. 26 The values obtained all indicate excellent windows and underscore the suitability for high-throughput screening.
GST-His6 AlphaScreen Counterscreen
We are aware that false positives often predominate the initial hit lists that arise from HTS campaigns. We are also aware that the AlphaScreen technology is not insensitive to this—hence our implementation of a GST-His6 counterscreen. 27 This AlphaScreen-based assay makes use of a His6-tagged GST protein, which brings the Ni2+-chelate acceptor and glutathione donor beads in proximity without relying on a protein-protein interaction. True inhibitors/stimulators of IN dimerization should not be scored as hits in this assay. On the other hand, compounds that were initially picked up because they interfere with the AlphaScreen technology itself (quenchers) will emerge as hits again in this counterscreen and can subsequently be removed.
Before including this assay in our workflow, we performed a pilot run with a control plate to evaluate the performance of our assay in a high-throughput setting. This plate had 32 positive control wells containing 100 µM bromophenol blue (absorbs emission light of 520-620 nm from acceptor beads) and 32 negative control wells containing 0.4% DMSO. Results are shown in Figure 7 , together with a calculated Z′ factor and SSMD of the assay. The resulting Z′ = 0.90 and β = −30.4 demonstrate an excellent suitability for HTS.
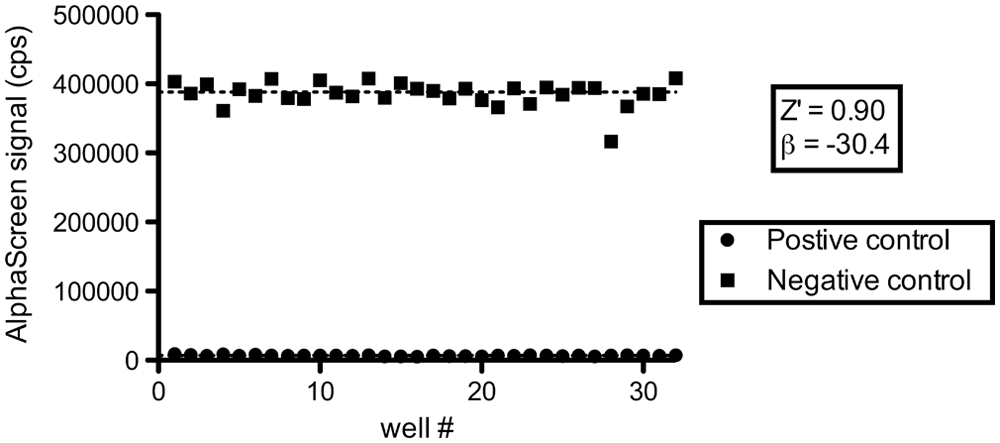
Glutathione S-transferase (GST)–His6 counterscreen quality control. A test plate for the GST-His6 AlphaScreen counterscreen, containing 32 positive (100 µM bromophenol blue) and 32 negative (0.4% DMSO) controls was prepared. Resulting signals were plotted and statistics calculated. Again, both Z′ factor and strictly standardized mean differences indicate an excellent screening window and strong controls for high-throughput screening.25,26 A single negative control outlier was not included in the calculations.
DTT and IN Dimerization
In their HTRF-based assay, Tsiang et al. 18 detected an inhibitory effect of reducing compounds, most notably DTT, on IN dimerization. We verified whether IN and DTT would behave similarly in our AlphaScreen-based assay and determined an EC50 of 2.19 mM for DTT (95% CI, 1.99–2.45 mM; Fig. 8 ). Simultaneously, we controlled for possible signal quenching due to DTT’s oxygen scavenging potential by performing the same titration in the GST-His6 counterscreen. In this assay, a much higher EC50 of 65 mM was found (54–79 mM). At 2.19 mM, quenching by DTT only accounts for ±3% of the total signal inhibition, demonstrating the inhibitory effect of DTT to be specific for IN dimerization. The EC50 determined in the AlphaScreen-based IN dimerization assay is in perfect agreement with the 2.2 mM reported by Tsiang et al. 18 Curiously, when DTT was diluted beyond 0.2 mM, the AlphaScreen signal of both IN dimerization and GST-His6 assays started to decrease as well. We ascribe this effect to the localized concentration and aggregation of GST-tagged protein on the surface of the glutathione donor beads when the environment is not sufficiently reducing. This could exclude the proteins from interaction with their partners (IN-His6 or Ni2+-chelate on the acceptor beads) and hence decrease the output signal.
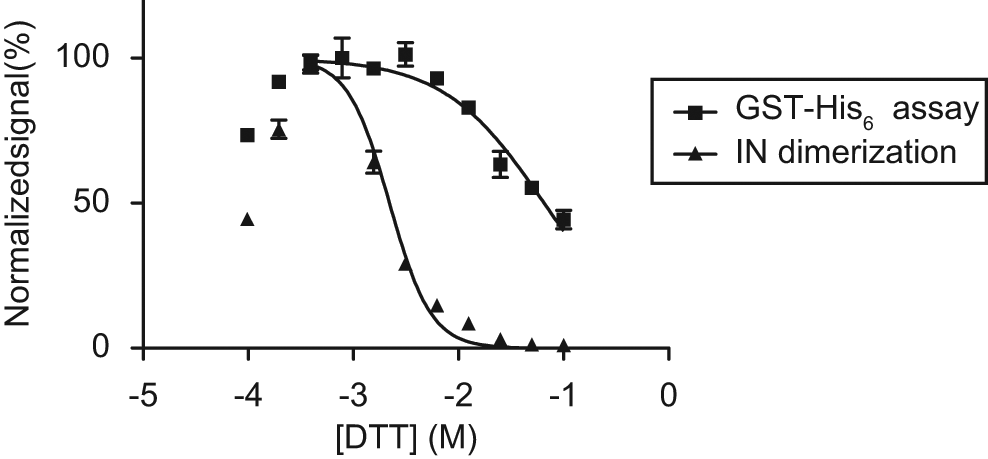
Effect of dithiothreitol (DTT) in the dimerization and glutathione S-transferase (GST)–His6 assays. We corroborated the inhibitory effect of the reducing agent DTT on integrase (IN) dimerization, as observed by Tsiang et al., 18 by titrating the compound in both dimerization and GST-His6 assays. Respective EC50 values of 2.19 and 65 mM were determined, demonstrating that the effect of DTT in the dimerization assay is not a result of DTT’s oxygen scavenging capacity and hence quenching during AlphaScreen signal generation. If DTT was reduced below 0.2 mM, the signal dropped equivalently in both assays.
Discussion
We report on the development of an AlphaScreen-based assay for the discovery of novel allosteric HIV-1 IN inhibitors. Compounds that inhibit or stimulate IN dimerization can now be identified in a robust high-throughput setting. As IN requires a dynamic equilibrium between at least dimers and tetramers for successful completion of its catalytic activities, any compound interfering with this precarious balance presents a likely inhibitor. 13 Proof of principle for this inhibitory strategy has previously been reported, generally through the use of peptides.13–16,20–22 In our workflow, we included a GST-His6 AlphaScreen counterscreen to rapidly and confidently purge initial hit lists of false positives.
Tsiang et al. 18 detected an inhibitory effect of reducing compounds, DTT in particular, on IN dimerization by using an HTRF-based assay. We here corroborated this effect and obtained a similar EC50 of 2.19 mM for DTT ( Fig. 8 ). These independent but highly similar results strengthen the confidence in both assays and underscore their validity for measuring IN dimerization, despite major mechanistic differences between both assay technologies. As is the case in the HTRF-based assay, we cannot fully exclude that only IN dimers contribute to signal generation. Theoretically, IN tetramers and higher order oligomers containing both GST- and His6-tags may also bring both beads together and contribute to the AlphaScreen signal. However, under the assay conditions we have chosen (the presence of detergent, Mg2+, and a total IN concentration [GST-IN plus IN-His6] as low as 30 nM), their contribution should be negligible, even in the presence of a stimulator.17,18 As a consequence, this assay, in its current state, is not suited for the discovery of specific IN tetramerization inhibitors. On the other hand, all IN dimerization inhibitors should also inhibit the formation of the functional IN tetramer (a dimer of dimers) and perhaps of higher order oligomeric species.
Tintori et al. recently selected a panel of 25 compounds by in silico screening for IN dimerization inhibitors (Tintori, C.; Demeulemeester, J.; Franchi, L.; Christ, F.; Debyser, Z.; Massa, S. and Botta, M. submitted for publication, 2011). These molecules were evaluated using the IN dimerization assay and GST-His6 counterscreen, resulting in the identification of several active compound classes. The most potent compound also displayed activity in a subsequent ELISA measuring overall IN catalytic activity (3P and ST). It is important to note that this compound, which was first selected in the dimerization assay, also inhibited integration as measured by a standard ELISA-based assay for catalytic activity. This again underscores the potential of this approach for the discovery of novel allosteric IN inhibitors.
In a recent study of the IN CCD dimerization interface, Serrao et al. applied the AlphaScreen-based assay described here to investigate the dimerization potential of various HIV-1 IN mutants (Serrao, E.; Thys, W.; Demeulemeester, J.; Al-Mawsawi, L.Q.; Christ, F.; Debyser, Z. and Neamati, N. submitted for publication, 2011). They identified a symmetrical “dimerization hot spot” to be critical for dimerization and catalytic activity. This site appeared to have a very low tolerance for substitutions (both conservative and nonconservative), hinting at a potentially high genetic barrier for the virus to develop resistance against inhibitors binding here. The results further encourage drug discovery efforts for IN dimerization inhibitors.
Because the binding site for LEDGF/p75 is located across the IN monomer-monomer interface, involving amino acid residues of both monomers, addition of this cofactor may modulate IN oligomerization. In fact, LEDGF/p75 and several of its derived peptides have been shown to promote IN tetramerization in vitro.13,28 The structure of LEDGIN 7, a small-molecule inhibitor of the IN-LEDGF/p75 interaction and our stimulatory positive control, mimics amino acids 365 to 368 of LEDGF/p75. 12 Hence, it comes as no surprise that our analysis of the IN dimerization assay seems to indicate that LEDGINs may affect the dynamic oligomerization equilibrium of IN. However, further investigation into their mode of allosteric inhibition of IN activity is required.12,29 In addition, the impact of the A128T IN resistance mutation and the comparable potencies found for LEDGIN 7 in the dimerization, the IN-LEDGF/p75 interaction, and the MTT/MT4 viral replication assays support the biological relevance of the assay. 12 The EC50 value determined for the INH5 interfacial peptide (4.5 µM) is more difficult to compare because of the large divergence in activities reported in the literature (from 60 nM to 88 µM). We would attribute these discrepancies mainly to the different assay formats used and potentially to varying reagent (peptide) quality.14,17 In any case, the potency of INH5 determined in our dimerization assay falls nicely within the range of reported values.
Aforementioned results corroborate that screening with the AlphaScreen-based IN dimerization assay and its GST-His6 counterscreen presented here can lead to the discovery of novel HIV integrase inhibitors. These allosteric IN dimerization modulators (INDIMs) are likely devoid of any cross-resistance with INSTIs, backing their potential as true next-generation IN inhibitors.
Footnotes
Acknowledgements
We thank Nam Joo Van der Veken and Martine Michiels for excellent technical assistance.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This work was supported by the Agency for Innovation by Science and Technology (CellCoVir, Strategic Basic Research [IWT-SBO] grant number 60813), the K.U. Leuven Research Council (grant number OT/09/047), the Research Foundation–Flanders (FWO, grant number G.0530.08), and the European Commission (Targeting HIV Integration Co-factors, THINC, FP7, grant HEALTH-F3-2008-201032). JD is a doctoral fellow for the FWO, and FC is funded by the Industrial Research Fund (IOF).