
Abstract
This article describes the development of a simple and robust fluorescence polarization (FP)–based binding assay and adaptation to high-throughput identification of small molecules blocking dsRNA binding to NS1A protein (nonstructural protein 1 from type A influenza strains). This homogeneous assay employs fluorescein-labeled 16-mer dsRNA and full-length NS1A protein tagged with glutathione S-transferase to monitor the changes in FP and fluorescence intensity simultaneously. The assay was optimized for high-throughput screening in a 384-well format and achieved a z′ score greater than 0.7. Its feasibility for high-throughput screening was demonstrated using the National Institutes of Health clinical collection. Six of 446 small molecules were identified as possible ligands in an initial screening. A series of validation tests confirmed epigallocatechine gallate (EGCG) to be active in the submicromolar range. A mechanism of EGCG inhibition involving interaction with the dsRNA-binding motif of NS1A, including Arg38, was proposed. This structural information is anticipated to provide a useful basis for the modeling of antiflu therapeutic reagents. Overall, the FP-based binding assay demonstrated its superior capability for simple, rapid, inexpensive, and robust identification of NS1A inhibitors and validation of their activity targeting NS1A.
Introduction
Influenza viruses 1 cause annual epidemics that are a major public health problem, causing approximately 36 000 deaths annually in the United States. Periodic pandemics result in much higher human deaths tolls. The worst influenza pandemic was in 1918–1919, killing approximately 50 million people worldwide, including an estimated 675 000 deaths in the United States. They are RNA viruses composed of three general types: influenza A, influenza B, and influenza C. Type A, influenza virus, which is responsible for these pandemics, has its reservoir in wild birds and infects multiple species, including a wide variety of warm-blooded animals, such as humans, swine, horses, dogs, cats, and other mammals. In contrast, type B influenza viruses are isolated almost exclusively from humans.
One candidate for a virulent pandemic influenza A virus is the H5N1 subtype, which was transmitted from birds to humans in 1997 in Hong Kong, resulting in six deaths among 18 infected persons. The H5N1 subtype also caused disease outbreaks in poultry in Asia, Europe, and Africa during late 2003 and early 2004. Although the ability to transmit H5N1 variants between human beings has been limited, there have been increasing concerns that H5N1-subtype influenza A viruses may develop the capacity for human-to-human transmission, potentially creating a pandemic strain. In addition, we are now in the midst of a pandemic caused by a virus originating in swine, the 2009 H1N1 virus, or “swine flu.” 2
Although vaccination remains the first choice for controlling viral activity, researchers and companies are searching for novel anti-influenza drugs to complement vaccines and to act as a first line of defense while vaccines are produced. There are currently two classes of antiviral drugs3,4 that are available for the prophylaxis or treatment of influenza: the adamantane-based drugs or M2 inhibitors targeting the viral ion channel and the neuraminidase inhibitors (e.g., oseltamivir). However, the clinical use of these inhibitors is limited by side effects, including renal and central nervous system symptoms.5,6 A more serious problem, though, is that small numbers of viral mutations lead to resistance, and because influenza-encoded RNA polymerase complexes have no proofreading activity, high mutation rates (ranging from approximately 1 × 10–3 to 8 × 10–3 substitution per site per year) 7 lead to the accumulation of multiple point mutations during replication. Thus, the frequent use of anti-influenza drugs is rapidly expanding resistance mutations in natural strains of the virus worldwide.8,9 The need to identify new targets for influenza drugs, especially targets that might control the H5N1 subtype, is therefore critical.
Since the early 1990s, scientists have found that multifunctional protein 1 (NS1) from influenza is important in a variety of processes during viral infection. 10 NS1 binds to both RNAs and proteins, interferes with host mRNA processing, controls viral RNA synthesis, regulates viral mRNA translation, and inactivates the host immune/ apoptotic response. Because of its critical functions at multiple levels of viral infection, the NS1 protein has been identified as a viable therapeutic target.11–13 In particular, its N-terminal RNA-binding domain (RBD; corresponding to the first 73 amino acids) nonspecifically binds to a wide range of double-stranded RNA (dsRNA) sequences, 14 sequestering viral RNAs and preventing activation of the 2′-5′ oligo(A) synthetase. Most importantly, though, NS1A is highly conserved across influenza A strains, 15 including the potentially deadly H5N1 strain. For these reasons, small molecules that antagonize NS1A and in particular disrupt dsRNA interactions could serve as potentially useful new antiviral agents.
Fluorescence polarization (FP) is a spectroscopic technique that monitors the rotational rate of a fluorophore using polarized emission filters. Because FP is homogeneous, rapid (mix and read), noninvasive, nonradioactive, relatively insensitive to pH changes, and independent of total fluorescence, this robust method is frequently employed in inhibitor screening assays.16,17 The method compares the tumbling rate of a labeled small molecule in complex with a large protein ligand to its rate when displaced by a competing ligand. Thanks to the development of multifunctional plate readers capable of high-throughput FP measurement (Perkin Elmer EnVision, Biotek Synergy, BMG PHERAstar, and Tecan Infinite), the application of this technique to drug discovery has grown explosively over the past decade.18–21 FP technology has a decided advantage over other screening methods that have been reported for identifying leads against NS1A because it does not use radiation 11 and directly probes molecular interactions, unlike cell-based assays.12,13
In this report, we sought to develop a simple, robust, inexpensive, and scalable FP screen that directly targeted NS1A and its interaction with dsRNA. The FP assay was carried out with 446 clinically proven chemicals in a 384-well format, and processing (sample preparation, liquid handling, and reading) was completed in less than 30 min per plate, excluding 1 h of compound incubation time. An initial screen identified six compounds, and subsequent assays and counterscreens led to a single, validated hit for further lead optimization. Because the hit likely binds to the active site of NS1A RBD, this work paves the way for lead optimization in concert with structure-based drug design.
Materials and Methods
Materials
Full-length (1–215) glutathione S-transferase (GST)–tagged NS1A protein, histidine (His)–tagged NS1A RBD (1–73) of NS1A/UDORN/72, and their mutants were prepared as described previously.14,22 The RNAs used for the FP assay were purchased from Integrated DNA Technologies (Coralville, IA) and prepared by annealing two 16-nucleotide single-stranded RNAs (ssRNAs) 23 at 90 °C for 2 min: (1) FAM-CCAUCCUCUACAGGCG (sense) and (2) FAM-CGCCUGUAGAGGAUGG (antisense). Ethidium bromide (EB), DMSO, epigallocatechine gallate (EGCG), and Tween-20 were purchased from Aldrich (St. Louis, MO), and yeast tRNA was obtained from Ambion (Austin, TX).
General methods
All fluorescence measurements were carried out in a 384-well, low-volume, round-bottomed, black polypropylene microplate (Nunc, catalog No. 267461) using an EnVision 2103 multilabel reader (Perkin Elmer, Waltham, MA) with excitation at 485 nm (BW = 14 nm) and emission at 535 nm (BW = 40 nm). Filters with excitation at 525 nm (BW = 45 nm) and emission at 595 nm (BW = 60 nm) were used exclusively for the EB displacement assay.
NS1A FP-binding assays
Fluorescein-labeled dsRNA (FAM-dsRNA) was prepared by combining sense and antisense ssRNAs in a hybridization buffer (50 mM Tris-HCl, pH 8.0, 50 mM KCl), incubated at 90 °C for 2 min and then cooled to room temperature for 1 h. For each assay, target protein binding with FAM-dsRNA was measured in a binding buffer (50 mM Tris-HCl, pH 8.0, 50 mM KCl, 1 mM DTT, 50 ng/µL tRNA, 0.02% Tween-20). In the binding assay, 10 µL of FAM-dsRNA was titrated with 10 µL of increasing concentration of NS1A proteins to calculate the dissociation constants (K d ) of NS1A, NS1A RBD, and mutant proteins. Competition assays with unlabeled dsRNA were conducted by filling each well with 10 µL of NS1A-GST (250, 500, and 1000 nM) and FAM-dsRNA (10 nM) mixture in a binding buffer and 10 µL of increasing concentrations of unlabeled dsRNA (0–80 nM). DMSO tolerance was measured by adding various concentrations of DMSO to a solution of NS1A-GST and FAM-dsRNA at final concentrations of 300 nM and 5 nM, respectively. In addition, DMSO titration was examined against FAM-dsRNA and FAM-ssRNA, respectively. For all FP-binding assays, fluorescence intensity (FI) and FP were measured after 1 h of incubation at room temperature. FP values were reported in millipolarization units (mP). The data were normalized according to ΔmP of negative and positive controls and fitted using nonlinear regression in Sigma Plot 11.0 (Systat Software Inc., San Jose, CA). All experiments were performed in duplicates, and data were averaged.
High-throughput screen and primary hit confirmation
A total of 10 nM FAM-dsRNA was mixed with 600 nM NS1A-GST in a twofold binding buffer. Ten microliters of dsRNA:NS1A-GST complex was added to assay-ready plates with a JANUS automated liquid-handling workstation (Perkin Elmer). Compounds dissolved in 100% DMSO at 10 mM concentration were first diluted in water to a final concentration of 100 µM, then 10 µL aliquots were transferred to assay-ready plates. Compounds from the National Institutes of Health (NIH) clinical collection (BioFocus DPI, San Francisco, CA) were screened at a final concentration of 50 µM. Data acquired from screening were mined for hit identification using the CDD database (Collaborative Drug Discovery, Burlingame, CA). Hit compounds identified from the primary screen were confirmed by titrating hit compounds (0–25 µM) with the same screening condition and counterscreened in parallel. Counter binding assay was conducted following the same protocol used in the FP-based binding assay excluding the NS1A-GST protein target. Specifically, hit compounds were diluted in a binding buffer as described above, and 10 µL of serially diluted chemical compounds was added to each well containing 10 µL of FAM-dsRNA:NS1A-GST complex or FAM-dsRNA.
EB displacement assay
EB displacement assays were performed to evaluate nonspecific binding characteristics of small chemical compounds with oligonucleotides. This assay was modified from a protocol described by Rishi et al. 18 Instead of hairpin DNA, unlabeled 16-nucleotide dsRNA, which has the same sequence used for the FP assay, was used. In detail, hit compounds identified from primary screening were diluted in hybridization buffer and were added to a mixture of EB and unlabeled dsRNA diluted in hybridization buffer. The final concentrations of EB and unlabeled dsRNA were 0.5 µM and 1 µM, respectively, whereas various concentrations of hits were evaluated. Test mixtures were incubated at room temperature for 30 min in PCR tubes and transferred to a black 384-well plate, and FI was monitored. All hit compounds were examined in duplicates with negative controls—(1) no inhibitor, (2) no RNA, and (3) no EB—to accurately verify displacement activity.
CD spectroscopy
Far-UV Circular Dichroism spectra were measured using an 815 CD spectrometer (Jasco Inc., Easton, MD) at 20 °C in a 1 mm quartz cuvette from 200 to 260 nm using a scanning rate of 100 nm/min with a 0.1 nm interval at the spectral band width of 2 nm and response time of 2 s. All spectra were corrected with corresponding blanks, and an average of three scans was obtained. The N73 (NS1A 1–73) polypeptide at a concentration of 10 µM in CD buffer (25 mM sodium phosphate, 25 mM sodium chloride, and 0.5 mM sodium azide at pH 6.5) was mixed with the compound (EGCG) in various concentrations in the CD buffer and incubated for 1 h at room temperature before scanning. Thermal stability was measured by heating from 6 °C to 80 °C with a heating rate of 1 °C/min. The change of ellipticity was recorded at 227 nm, and the results were expressed as Δε. Tm (midpoint of thermal transition) was analyzed using nonlinear regression fitted to a sigmoidal model.
Results
Development and Optimization of an FP-Based Binding Assay for NS1A
In this research, we first investigated whether a filter binding assay previously reported by Chien et al.
23
could be adapted to an FP assay (
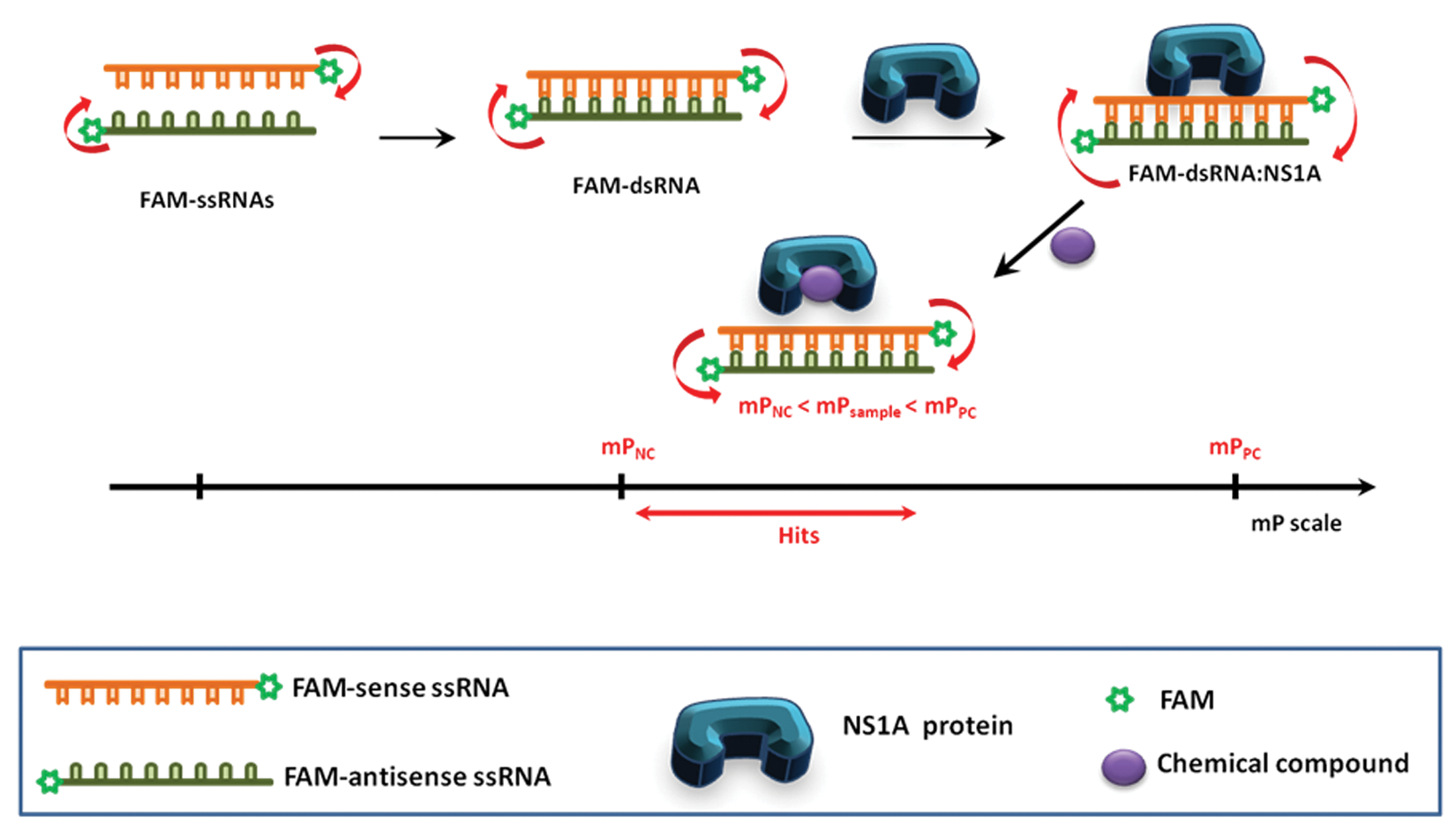
Schematic representation of the fluorescence polarization (FP)–based assay monitoring biological interaction. The scale bar represents arbitrary comparison of FP values. If a chemical compound displaces the FAM-dsRNA from the NS1A, the FP decreases to mPNC. Any chemical compound that induces mP decrease compared with mPPC (red line on the mP scale) will be identified as hits.
To demonstrate that FP is independent of the total FI, 17 and to determine the optimal concentration of the FAM-dsRNA probe, the FP and FI of FAM-dsRNA were determined over the range of 0 to 20 nM. Figure 2A shows that FP values were stable at concentrations as low as 5 nM FAM-dsRNA with less than 5% of coefficient variation (CV = 100 × standard deviation/mean), whereas the absolute FI values were proportional to FAM-dsRNA concentrations, as expected. In consequence, 5 nM was chosen as the optimum concentration of FAM-dsRNA for all further experiments.
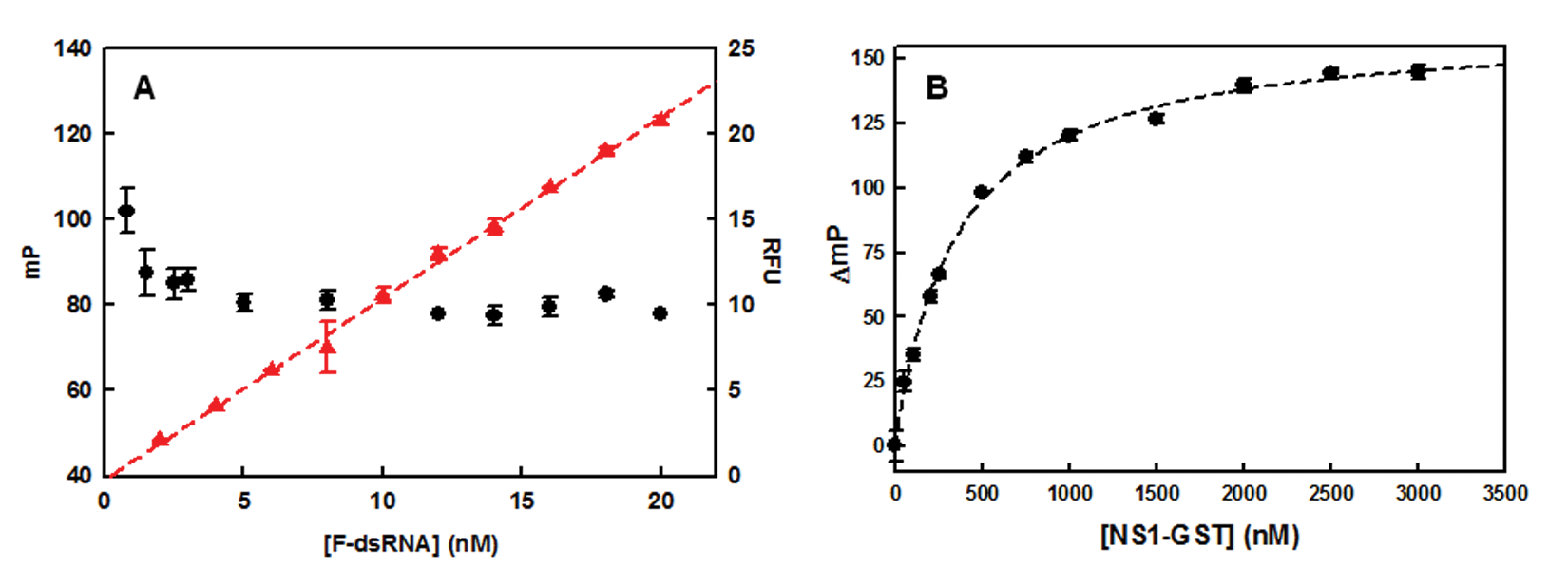
Fluorescence characteristics of the fluorescence polarization (FP)–based binding assay. (
Next, two different versions of NS1A (N73 RBD tagged with His [N73-His] and full-length N-215 tagged with GST [NS1A-GST]) were assayed with the dsRNA by FP. Because the GST tag was used in a previous binding assay, 23 we hypothesized that this construct might not only be functional but also could maximize the mass difference between dsRNA and complex, resulting in a larger change in FP. Free GST was assayed with FAM-dsRNA in parallel as a control. As shown in Figure 2B , NS1A-GST (MW ~56 000 Da) showed a dose-dependent increase in FP (K d = ~200 nM and Hill coefficient = ~1) and had a predicted wide dynamic range of ~120 ΔmP. The K d determined using equations suggested by Roehrl et al. 24 (Q = ~1.2, Q = FI of bound FAM-dsRNA/FI of unbound FAM-dsRNA) was ~160 nM, which is comparable with one that was obtained by nonlinear regression fit. GST alone showed no change in signal (data are not shown). In contrast, N73-His exhibited about 100-fold weaker binding affinity (calculated K d was ~25 µM). This result accords with an earlier study 23 that showed dsRNA binds more weakly to NS1 RBD than to full-length NS1A. The GST-tagged construct was therefore chosen for assay development.
To ensure that background with the FP assay would be low, we assessed the extent to which false signals might be observed. Structural and mutational analyses showed that Arg38 of the RBD is essential for dsRNA binding.14,25 Therefore, to determine whether there was a high specificity for the protein target, we assayed a mutant NS1A protein tagged with GST (R38A-GST, mutating Arg38 to Ala 14 ) and found it did not yield an FP signal. Because tRNA acts as a sink for nonspecific RNA binders, we examined the polarization of the FAM-dsRNA:NS1A-GST complex in the presence and absence of 50 ng/µL tRNA. Transfer RNA did not yield changes in FP signals or K d (data are not shown). The influence of incubation time on signal variation was also examined. Neither FP nor FI signals changed significantly during 4 h of incubation at room temperature or after 24 h at 4 °C. Based on these results, we decided to use GST-tagged NS1A protein in the presence of 50 ng/µL tRNA in the assay and measured fluorescence characteristics after 1 h incubation time at room temperature.
Detergents are frequent additives in high-throughput screen (HTS) campaigns. They are used not only to prevent protein absorption to the polypropylene wells but also to minimize the interfering effect of promiscuous inhibitors acting via colloidal aggregate formation. As studied thoroughly by Shoichet’s group,26,27 detergents significantly block aggregator-based inhibition, but the sensitivity of compound aggregation varies per compounds and assay conditions. Although moderate concentrations in the range of 0.015 to 0.1% of such detergents have been commonly applied in various HTS campaigns, it may be difficult to eliminate all potential aggregator-sensitive inhibitors. Because immune compounds may become incorporated into micelles when the detergent concentration is higher than its critical micelle concentration (CMC of Tween 20 = ~0.007% w/v), we used mP to examine the effect of Tween 20 (0%−0.1% [w/w %]) on the binding affinity of NS1A and FAM-dsRNA complex. Tween 20 was found not to alter the mP up to 0.1%. In addition, dose-dependent binding of FAM-dsRNA in a serial dilution of NS1A was tested with 0.02% of Tween 20 and without Tween 20, and we did not observe any distinctive difference in mP signal (data are not shown). To efficiently eliminate any potential aggregator-based inhibitions and to improve the precision of the liquid-handling process, 0.02% was chosen as a final concentration of Tween 20.
Assay Validation
As a surrogate for a small-molecule ligand, a displacement assay was carried out with unlabeled dsRNA. Unlabeled dsRNA and double FAM-labeled dsRNA were prepared separately by denaturing at 90 °C for 2 min followed by cooling to room temperature. Unlabeled dsRNA (0–80 nM) was then titrated with FAM-dsRNA (10 nM) and NS1A-GST complex at three different concentrations of NS1A-GST (100, 200, and 300 nM). Because both unlabeled and double FAM-labeled dsRNAs duplexes are thermodynamically stable at room temperature (Tm = 59 °C; http://www.basic.northwestern.edu/biotools/oligocalc.html), strand displacement events leading to the formation of heterogeneous duplexing was assumed to be so unlikely as to have a negligible effect on measured mP. As shown in Figure 3A , FP values decreased as unlabeled dsRNA displaced FAM-labeled dsRNA from the complex and released free FAM-labeled dsRNA in a concentration-dependent manner. The lowest FP under the large excess of unlabeled dsRNA was ~90% of FP of unbound FAM-dsRNA. As expected, the dynamic range was greater at higher concentrations of NS1A-GST ( Fig. 3A ). However, the degree of FP variation in each plot after normalization ( Fig. 3B ) appeared to be uniform because FAM-dsRNA was not saturated in this concentration range. Moreover, different protein concentrations exhibited comparable IC50 values (26–35 nM, calculated using nonlinear regression fitted to a logistic model). Ultimately, 300 nM (70% binding) was chosen as the NS1A-GST concentration for the assay, following Lokesh et al., who suggested that the ratio between the receptor (NS1A-GST) concentration and the K d of the ligand (FAM-dsRNA) should be at least 1. 19
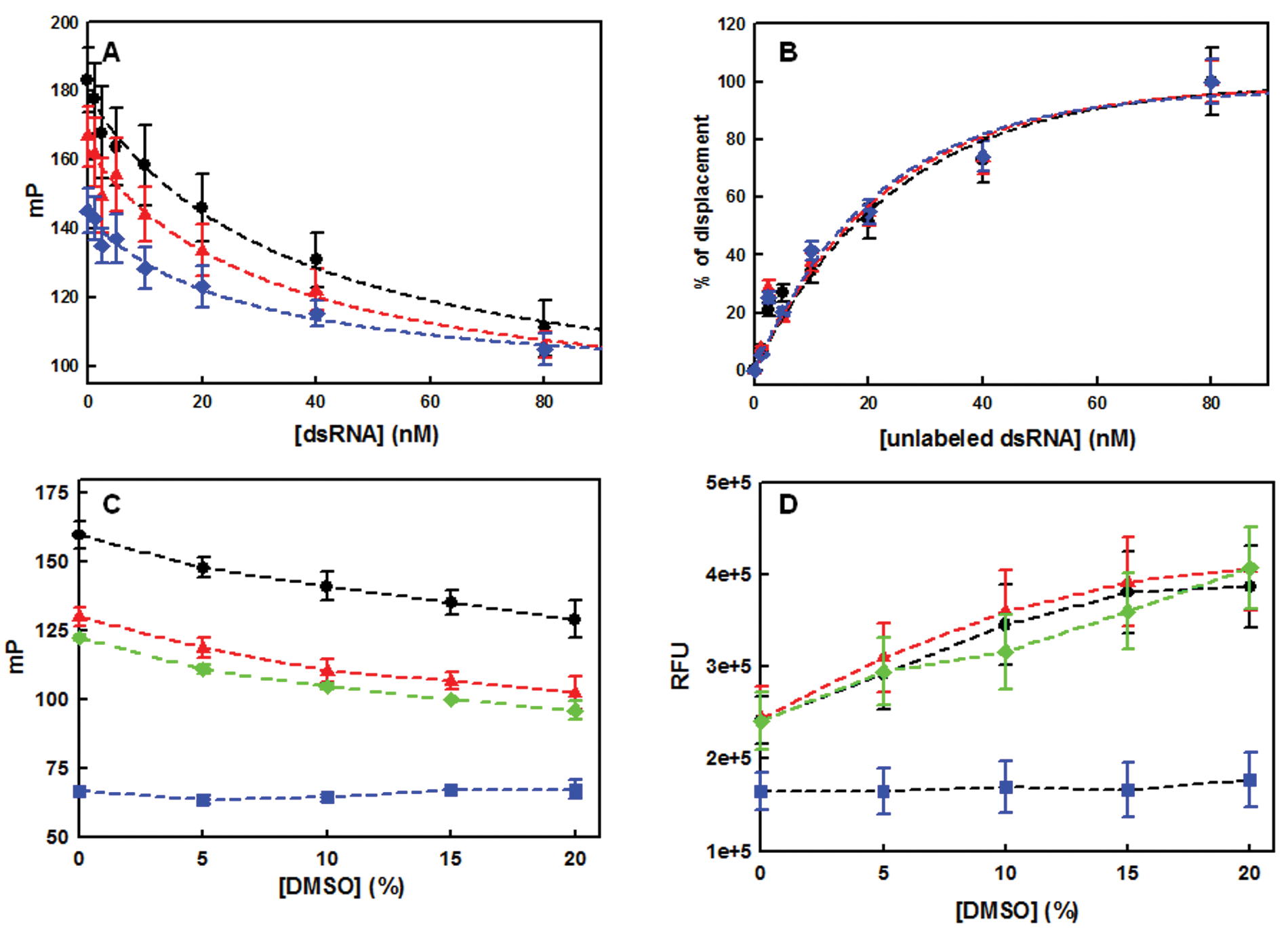
Assay validation. (
Given that commercial chemical libraries are normally dissolved in 100% DMSO, the influence of this solvent on NS1A-GST:dsRNA interactions was also addressed. Protein:FAM-dsRNA complexes were prepared with NS1A-GST and N73-His, and both were titrated with DMSO in the range of 0% to 15% (w/v). In addition, the DMSO modulation of signals from free FAM-dsRNA and FAM-ssRNA was assessed as a negative control. At greater than 1.5% (w/v) DMSO, the FAM-dsRNA:protein complexes and FAM-dsRNA exhibited FP value reductions, whereas the FI value increased in proportion to the DMSO concentration. In contrast, the FP and FI values of FAM-ssRNA remained constant ( Fig. 3C , D ). These results suggested that DMSO likely denatured the dsRNA rather than directly obstructing the binding of dsRNA to protein. Both the FP and FI values of FAM-dsRNA were stable at less than 1.5% DMSO, a reasonable concentration for many compound dilutions in the screening. This experiment also emphasized that both FP and FI values should be monitored to avoid false-positive hits that might arise due to RNA denaturation whether compounds are fluorescent or not.
Finally, assay validation ( Fig. 4A ) was carried out by filling half of an assay plate with a positive control (FAM-dsRNA:NS1A-GST) and the other half with a negative control (FAM-dsRNA), followed by an addition of 10 µL of DMSO diluted in binding buffer (the final DMSO content was 0.5%). FP and FI signals were measured, and the z′ factor 28 was then calculated based on FP values from three independent experiments. The average z′ factor was ~0.7, confirming that these assay conditions were robust for HTS.
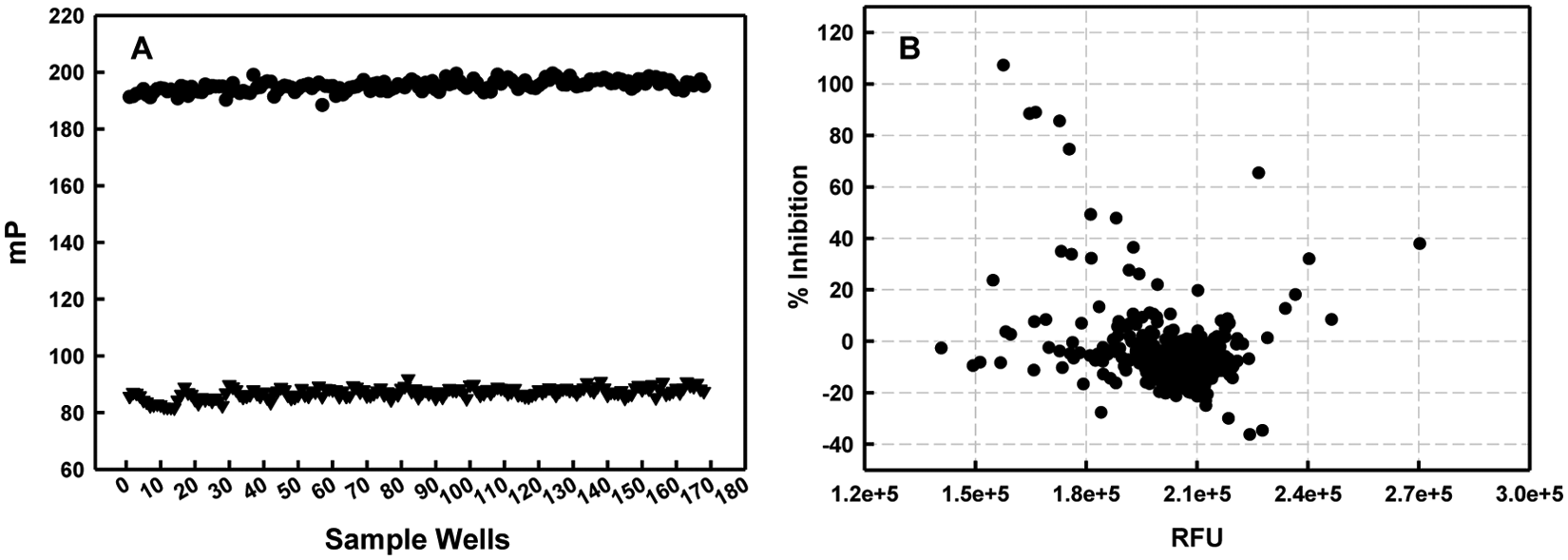
Typical plots of assay validation and primary screening. (
High-Throughput Primary Screening
Four hundred sixty-six compounds from the NIH clinical collection were plated into two 384-well plates (library 1 and library 2). A total of four plates (including duplicates) were then tested at 50 µM final compound concentrations. Simultaneous scanning of FI and FP was completed in less than 2 min per plate. In each screening plate, columns 1 and 2 (total of 32 wells) were used for positive controls, whereas columns 23 and 24 (total of 32 wells) were used for negative controls. Assay plates were revalidated after screening to ascertain the data reliability within a plate and across plates, as summarized in Table 1 . Signals from duplicated plates were very reproducible with large signal-to-noise (S/N) ratios (22.49–43.17), demonstrating the high quality of the signals. In particular, S/B ratios were generally greater than twofold and Z′ values were in the range of 0.71 to 0.82, again showing the excellent stability of the FP assay.29,30
Statistical Performance of Individual Screening Plates a
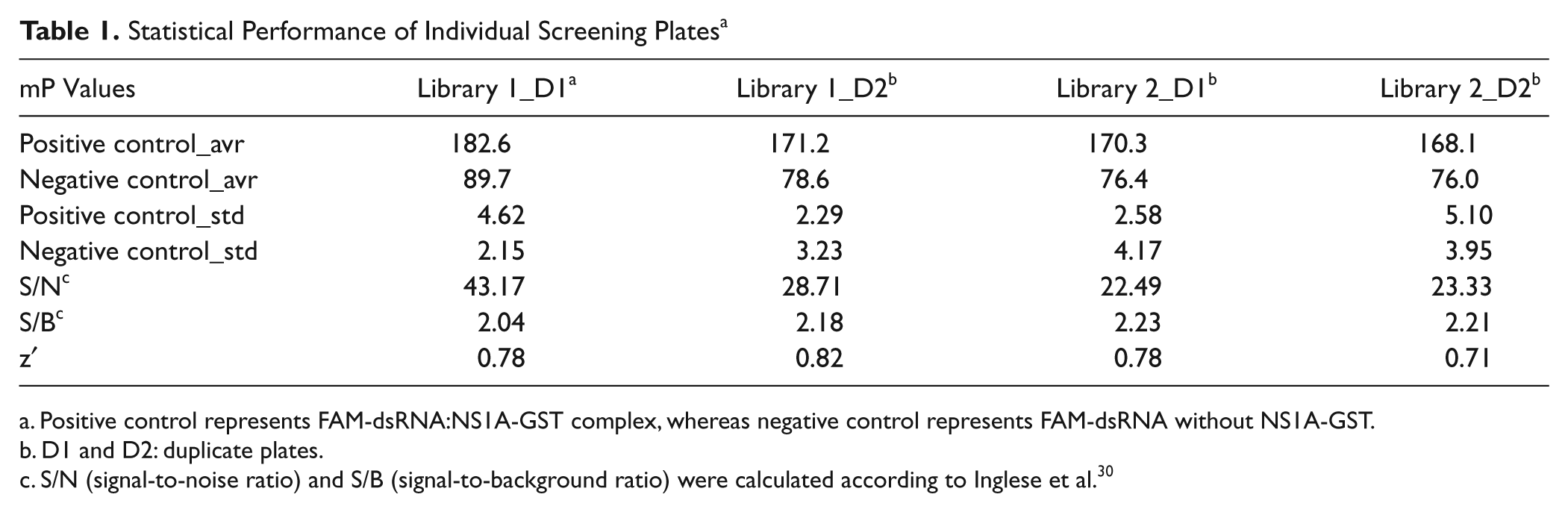
Positive control represents FAM-dsRNA:NS1A-GST complex, whereas negative control represents FAM-dsRNA without NS1A-GST.
D1 and D2: duplicate plates.
S/N (signal-to-noise ratio) and S/B (signal-to-background ratio) were calculated according to Inglese et al. 30
Typical data from the screen are illustrated in Figure 4B . The FP value of free FAM-dsRNA with DMSO (mPNC) corresponded to 100% inhibition, whereas the FP value of FAM-dsRNA:NS1A-GST with DMSO (mPPC) corresponded to 0% inhibition.
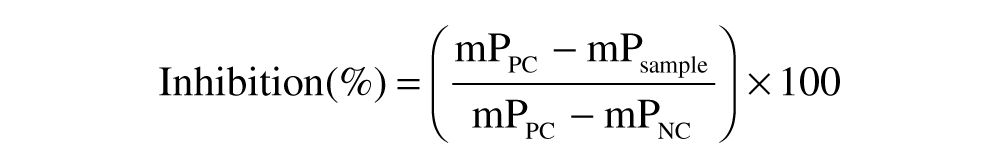
where mPPC, mPNC, and mPsample are the FP values of the positive control, the negative control, and the screening compound. If a compound gave greater than 50% inhibition based on its FP value, it was defined as a hit. To eliminate any false hits caused by nonspecific fluorescence interference, total FI was used as a second filter. 31 In this initial screen, compounds having more or less than 30% (a cutoff commonly employed in FP-based screening campaigns18,32) of total FI compared to the controls were considered as either enhancers or quenchers. The six compounds (SAM001246577, SAM001247031, SAM001246818, SAM001246816, SAM001246892, and SAM001246737) satisfying this hit criteria were identified as primary hits and further validated.
Hit Validation
Figure 5 represents various modes of interaction between small molecules and the dsRNA:NS1A-GST complex that might potentially affect FP values. The counterscreen was designed to assess chemicals interfering with dsRNA directly via intercalation (modes E and H in Fig. 5 ) or further denaturation (modes F, I, and K in Fig. 5 ). Counterscreening was carried out using the same binding assay protocol but excluded NS1A-GST, so that the parameters measured were the polarization of the dsRNA (negative control) in the presence and absence of compounds.
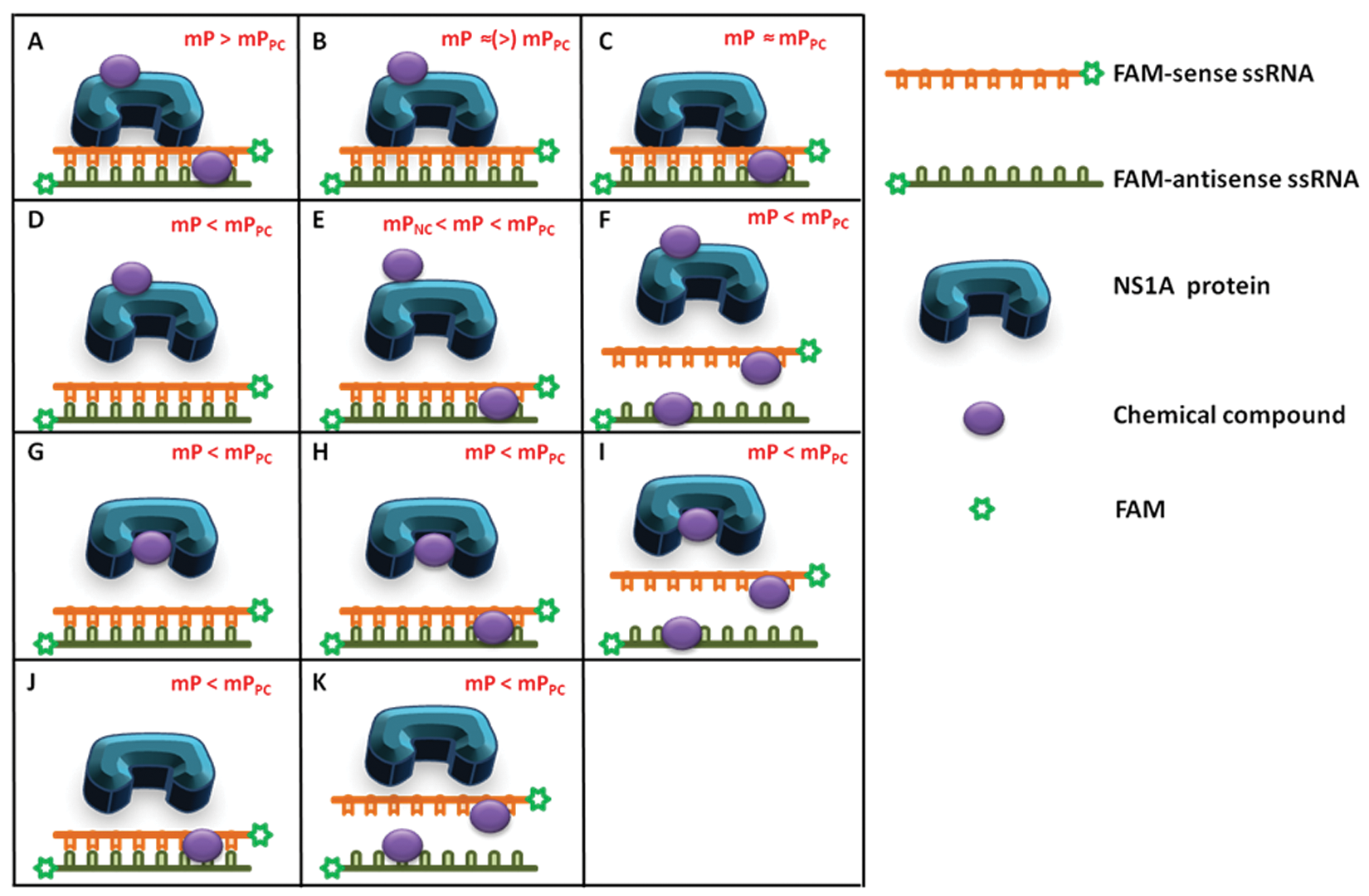
Possible modes of interaction for the small molecules in the fluorescence polarization (FP) binding assay. First, small molecules may interact only with dsRNA via intercalation (C, J, or K). This interaction may not affect FAM-dsRNA:NS1A binding (C), lead to the displacement of bound protein (J), or denature FAM-dsRNA (K). Modes J and K can produce mP values lower than mPPC and thus can be identified as false-positives. Second, small molecules may interact with both dsRNA and NS1A simultaneously (A, E, F, H, or I). Modes E, F, H, and I can be considered as false hits because they lead to the disruption of the dsRNA:NS1A-GST interaction and produce mP values lower than mPPC. Third, small molecules may interact only with NS1A through its binding pocket (G, true hit) or nonspecifically (B or D). It is possible that the nonspecific interaction of small molecules with NS1A will induce structural changes in this protein, displacing bound FAM-dsRNA and decreasing mP values lower than mPPC (D); this can also be considered a false-positive. Modes A, B, and C can be easily identified by primary screening because these interactions increase FP values higher than the positive control or similar; any other modes that reduce FP values will potentially yield misleading hits.
All compounds deemed to be hits in the initial screen were purchased from commercial vendors, freshly prepared in 100% DMSO, and diluted (0–25 µM) for rescreening ( Fig. 6A ) and counterscreening ( Fig. 6B ). Percentage mP change in Figure 6B represents the % ratio of mP with and without compound in FAM-dsRNA solution. If the extent of % mP decreased, this indicates that the compound may have denatured the dsRNA. On the other hand, if the extent of % mP increased, this suggests that the compound interacts primarily with dsRNA, although this assay is not sensitive enough to detect the level of FP increase caused by intercalation. Gratifyingly, no compound showed significant interactions with the RNA, as indicated in Figure 6B . That said, compounds SAM001246818 and SAM001246816 did not show reproducible inhibition, whereas compounds SAM001246577 and SAM001246737 exhibited lower than 40% binding at 25 µM. Compounds SAM001246892 and SAM001247031 reproducibly yielded binding as high as ~80%. Nonetheless, the inhibition activity of compound SAM001246892 was not saturated at the highest concentration of compound as shown in Figure 6A , and the apparent IC50 (~6 µM) was much higher than that of SAM001247031 (<1 µM). Compound SAM001246892 was also found to be a compound that frequently showed activity in other assays (45 active vs 232 inactive out of 297 assays tested) according to the Pubchem bioassay data bank (http://pubchem.ncbi.nlm.nih.gov/), suggesting the inhibition activity in this assay could be nonspecific. Contrariwise, SAM001247031 (EGCG) has been well known as a useful reagent for prevention and/or treatment of various life-threatening diseases, such as cancer, bacterial infection, HIV, and diabetes. 33 As EGCG showed dose-dependent activity in our FP assay with a nanomolar range of IC50 and was also recognized to be active in viral infection,34–36 we focused attention on compound SAM001247031.
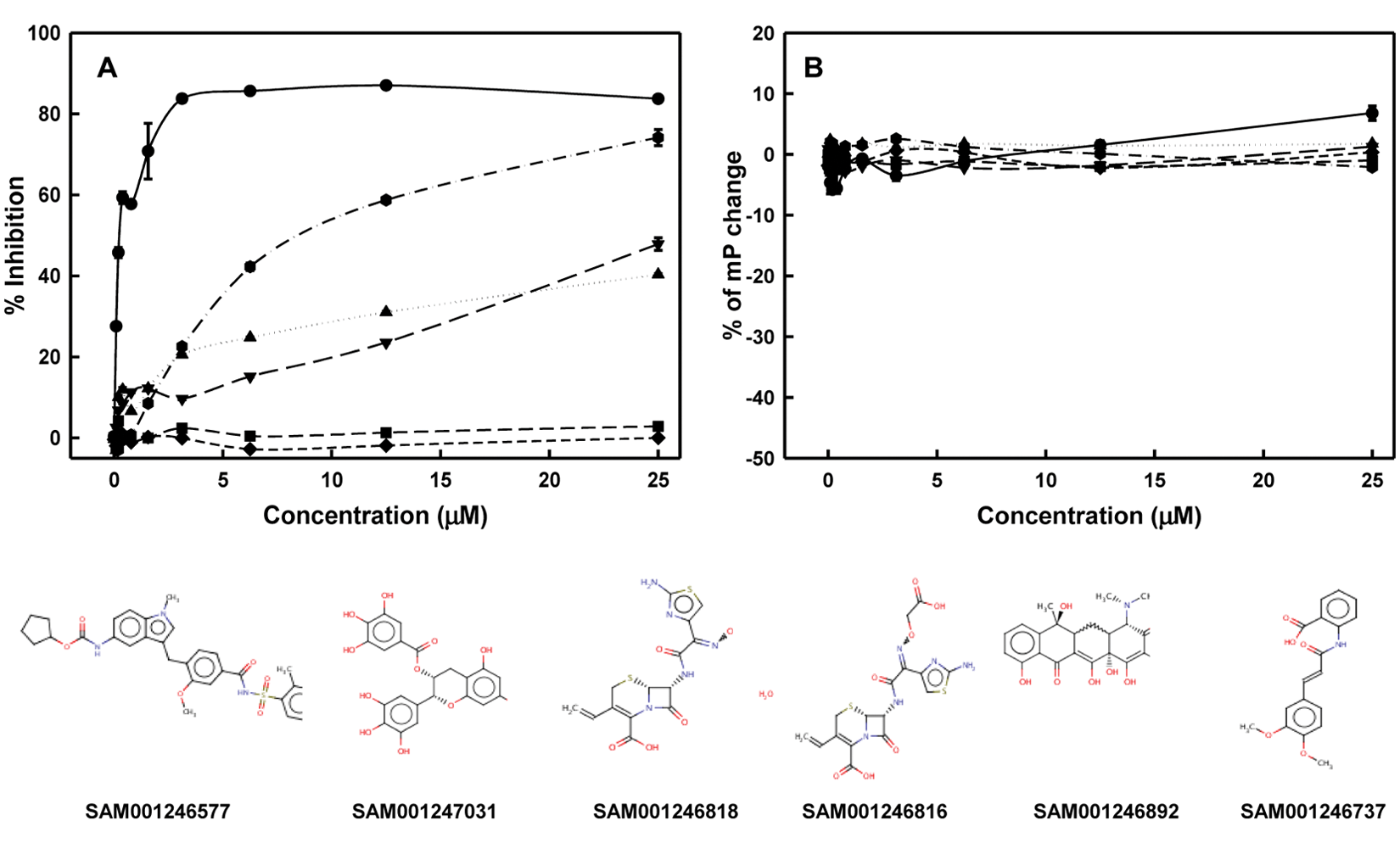
Dose-dependent response curves of hits obtained from the primary high-throughput screen and their chemical structures. (
We further investigated the inhibition mechanism of SAM001247031 to confirm its selective drug activity using an EB displacement assay. The EB displacement assay has been widely employed as a tool to rapidly evaluate the interaction between drugs and nucleic acids18,37 via intercalation or minor groove binding. This assay assumes that if a compound binds to dsRNA, it will displace prebound EB (K d = ~10−5 M), 37 which can in turn be measured by a decrease in the fluorescence of EB. Although the initial screen had already indicated that ECGC should not interact with dsRNA, the counterscreen was a necessary validating step because FI is more sensitive than FP in general. Therefore, SAM001247031 (EGCG) was assayed using the EB displacement assay. Because of the potential for nonspecific fluorescence quenching or enhancement by compounds at 530 nm excitation, EGCG was also tested against EB alone as a negative control. EGCG reproduced the FP-based dose-dependent response but showed no activity in the EB displacement assay over a concentration range from 0 to 4 µM, confirming that the mode of inhibition was specific (data are not shown.). The IC50 and Hill slope estimated using nonlinear regression fitted to a logistic curve were ~290 nM and ~0.8, respectively. The equilibrium dissociation constant (K i ) for the protein:drug complex was calculated to be 30.7 nM, using the equation described by Munson and Rodbard. 38
Characterization by Circular Dichroism Spectroscopy
CD spectroscopy is a powerful technique to study the structures of proteins in solution. Changes in CD signals in the presence of ligands can be used to determine the strength of the interaction and to provide information about the mechanism of action. Although full-length NS1A did not possess a unique secondary structure, the truncated form of NS1-RBD (N73) has been shown to possess a highly α-helical backbone structure while retaining all of the dsRNA binding and dimerization activities of the full-length protein. 39 Monitoring the change in the α-helical structure of N73 upon EGCG addition was therefore expected to further validate the mode of inhibition. Again, the mutant R38A was examined in parallel to monitor binding specificity.
The CD spectra of both purified wt N73 and mutant N73 dimers in the absence of EGCG were identical ( Fig. 7A ) as expected based on a previous study that showed Arg38 mutation does not affect its dimeric structure. 14 Both spectra exhibited minima at ~208 nm and 227 nm, a property that is characteristic of highly α-helical proteins. The CD spectra at 227 nm were used to monitor thermal stability from 6 °C to 80 °C with a dilution series of EGCG. Ellipticity was plotted as a function of temperature, and thermal denaturation curves of wt N73 ( Fig. 7B ) showed a Tm increase of 10 °C in the presence of 400 µM of EGCG. In contrast, no melting temperature changes were observed with mut N73 using the same dilution series of EGCG, although the free mut N73 showed similar Tm (41 °C) compared with the wild type. The inset in Figure 7B showed that the Tm of wt N73 increased proportionally to the concentration of EGCG, with saturation occurring at 400 µM of EGCG and higher concentrations of EGCG yielding decreased S/N ratios and noninterpretable scatter at higher temperatures. In accordance with the structural studies of NS1A RBD, this result implies that EGCG stabilizes the dimeric structure of N73 by specifically binding to the dsRNA binding site containing Arg38. Alternatively, specific binding of EGCG to a full-length NS1A can be validated by using isothermal titration calorimetry or differential scanning calorimetry, which measure thermodynamic characteristics as well as binding stoichiometry without relying on protein secondary structure.
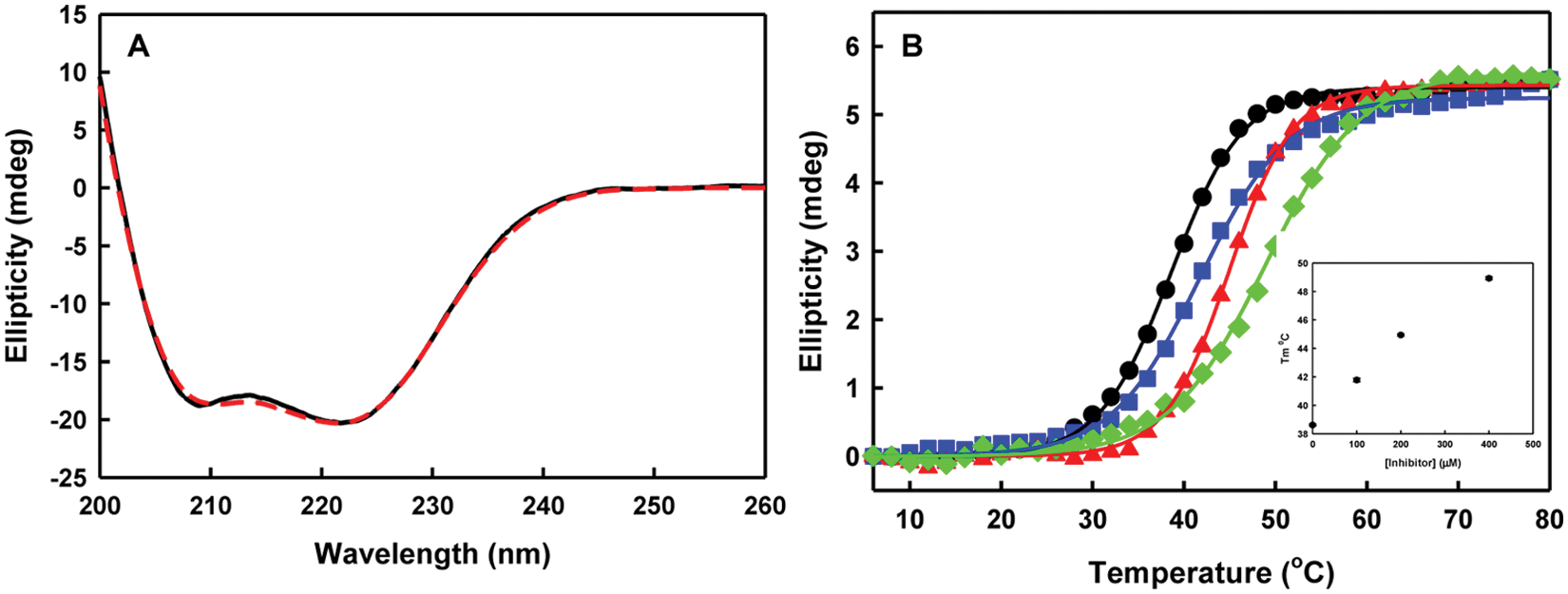
CD spectra and thermal stability of wt N73 and mut N73. (
Discussion
Influenza A viruses have been a major concern in public health because of their periodic pandemic threat. The highly conserved nonstructural protein of the influenza A virus (NS1A) binds to double-stranded RNA in host cells. Rapid evaluation of small molecules that can compete for binding to dsRNA is therefore an important strategy for antiflu drug development, including the potentially deadly H5N1 strain.
Several assays11–13 have been developed or adapted for high-throughput identification of small molecules that target NS1A. Basu et al. developed a yeast-based assay and identified four chemicals that phenotypically suppress NS1 function 12 and optimized those leads. 13 The resultant compounds blocked dsRNA-dependent activation of a transfected interferon-β promoter construct, but the mode of inhibition was uncertain because of the complicated nature of the cell-based system. A filter-binding assay measuring the specific interactions between recombinant His-NS1A and radiolabeled νRNA has also been adapted to a format suitable for automated screening by Maroto et al. 11 The authors identified three compounds that inhibited NS1A binding to νRNA at submicromolar concentrations, which reduced the cytopathic effects of the virus. Unfortunately, the authors did not disclose the active chemical structures. They also employed only one counterscreen with Staufen, 40 a protein that is known to have a high affinity for a variety of RNA molecules and whose relevance to dsRNA binding by the NS1A RBD is minimal. They used only one assay to discriminate nonspecific inhibitors, whereas more diverse assays are usually involved in early-stage drug screening.
We have developed a simple FP-based binding assay targeting the RNA binding domain of the NS1A protein and optimized this assay for high-throughput inhibitor screening. As a proof of principle, 446 clinically proven drugs were screened. Through stringent validation, the compound EGCG was confirmed to be active in the submicromolar range. This was gratifying because the antiflu activities of EGCG, a main ingredient of green tea extract and a family of catechine (polyphenolic compounds), had already undergone extensive investigation in a series of cell-based assays and animal models. Nakayama et al. 34 reported that EGCG agglutinates influenza virus, preventing it from being absorbed in MDCK cells. Song et al. 35 evaluated the potential of catechine derivatives and reported that EGCG and epicatechin gallate were most effective in inhibiting viral replication in MDCK cells. These compounds were effective with all influenza subtypes assayed. Furuta et al. 36 has also studied the effect of various hydroxyl substituents of EGCG on anti-influenza virus activity. These results confirm that the facile assay we have developed may have an increasing use for the rapid identification of compounds against pandemic influenza viruses.
The coupling of a molecular mechanism to antiviral properties is critical for drug development. A thermal denaturation assay showed that EGCG in fact bound to the dsRNA binding motif containing Arg38.
Our direct screening for a mechanism has therefore led to a compound that was already known to have a potent antiviral activity and is now revealed to have an interesting mode of action. This discovery is especially important because the structure of the dsRNA binding site is known, and thus structure-based design can potentially be used to further optimize this lead. In contrast, although compounds identified in other screens have claimed to inhibit the virus via the dsRNA-binding activity of NS1A, when we assayed the four compounds that were discovered in the more complex yeast cell-based assay, 12 none of them showed inhibition activity in our FP-based binding assay (data not shown).
Footnotes
Acknowledgements
We thank the National Institutes of Health (U01 A1074497), Welch Foundation (H-F-0032), and The Texas Institute for Drug and Diagnostic Development for support of this work. Special acknowledgments are given to Dr. Angel Syrett and Jin Hyung Lee for carefully reading this article.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.