
Abstract
The authors report here higher throughput screening (HTS) assays for the evaluation of CYP3A4 inhibition and CYP3A4 induction in human hepatocytes using a novel CYP3A4 substrate, luciferin IPA (LIPA). Using human recombinant CYP450 isoforms, LIPA was found to be metabolized extensively by CYP3A4 but not by CYP1A2, CYP2C9, CYP2C19, CYP2D6, or CYP2E1. In the 384-well plate CYP3A4 inhibition assay, the known inhibitors 1-aminobenzotriazole, erythromycin, ketoconazole, and verapamil were found to cause extensive (maximum inhibition of >80%), dose-dependent, statistically significant inhibition of LIPA metabolism. The non-CYP3A4 inhibitors diethyldithiocarbamate, quercetin, quinidine, sulfaphenazole, ticlopidine, and tranylcypromine were found to have substantially lower (maximum inhibition of <50%) or no apparent inhibitory effects in the HTS assay. In the 96-well plate induction assay, the CYP3A4 inducers rifampin, phenobarbital, carbamazepine, phenytoin, troglitazone, rosiglitazone, and pioglitazone yielded dose-dependent induction of LIPA metabolism, whereas the CYP1A2 inducers omeprazole and 3-methylcholanthrene did not display any induction in the CYP3A4 activity. The high sensitivity and specificity of the assays, the relative ease of execution, and reduced cost, time, and test material requirements suggest that the HTS assays may be applied routinely for screening a large number of chemicals in the drug discovery phase for CYP3A4 inhibitory and inducing potential.
Introduction
P
As a result of the serious outcomes of DDIs, the US Food and Drug Administration (FDA) requires clear definition of DDI potential of drug candidates for new drug applications (NDAs).7–9 Definition of DDI potential of a new drug includes investigation on the potential of the new drug to be either a perpetrator or a victim with existing drugs. The approaches to evaluate DDI potential have been systematically defined by the US FDA, with the major emphasis being placed on the evaluation of the interaction of the new drugs with cytochrome P450–dependent monooxygenases (CYP). The CYP family is represented by multiple isoforms, each with specific selectivity for certain chemical structures. Of the various isoforms, CYP3A (CYP3A4, CYP3A5) collectively represent the most abundant hepatic isoforms and are known to be involved in the metabolism of more than 50% of existing drugs.10,11 CYP3A4 has been identified as the most important P450 isoform involved in clinically significant DDIs. 12
The commonly used approach to measure CYP3A4 activity is the use of high-performance liquid chromatography accompanied with mass spectrometry (LC/MS) to quantify the metabolism of prototypical substrates such as testosterone, midazolam, and nifedipine. 13 Although the use of LC/MS analysis is a routine practice and higher throughput automated approaches are available, 14 it remains a bottleneck when a large number of samples are to be analyzed. We have recently reported that CYP3A4 activity in human hepatocytes can be readily quantified by ATP-luciferin-based luminescence using a multiwell plate reader in a rapid, highly sensitive, and highly quantitative manner. 15 We report here higher throughput, luciferin IPA (LIPA)–based human hepatocyte assays for the evaluation of two major mechanisms of DDI—namely, CYP3A4 inhibition and CYP3A4 induction. The assays reported here should complement the current higher throughput approaches for the evaluation of P450-related drug–drug interactions.16–18
Materials and Methods
Chemicals and reagents
All cell culture media and supplements and model P450 inhibitors (1-aminobenzotriazole, erythromycin, ketoconazole, verapamil, diethyldithiocarbamate, quercetin, quinidine, sulfaphenazole, ticlopidine, and tranylcypromine) were obtained from Sigma-Aldrich (St. Louis, MO). Model P450 inducers (rifampin, phenytoin, carbamazepine, and phenobarbital) were obtained from Sigma-Aldrich, whereas troglitazone, rosiglitazone, and pioglitazone were obtained from Cayman Chemicals (Ann Arbor, MI) as a part of PPAR-γ ligand pack. LIPA, luciferin standard, and luciferin detection reagent were obtained from Promega (Madison, WI).
Enzyme specificity assay
The specificity of the LIPA was screened using human recombinant P450 Cerosomes (CYP3A4, CYP2C19, CYP2E1, CYP2C9, CYP1A2, and CYP2D6; Premas Biotech, Gurgaon, India). A constant amount of cytochrome P450 (12.5 pmol/mL) was incubated with 1 µM LIPA and 1 mM NADPH for 20 min. All incubations were performed in 150 µL of 100 mM potassium phosphate buffer (pH 7.4). Following incubation, 50 µL of the reaction mixture was transferred to a white plate to which 50 µL of luciferin detection reagent was added.
Human hepatocytes
The human hepatocytes used in this study were isolated and subsequently cryopreserved in our laboratory using our previously published methods for hepatocyte isolation and cryopreservation19–21 from nontransplantable human livers made available for use in research. Cryopreserved human hepatocytes pooled from 10 donors were used for the high-throughput screening (HTS) inhibition assay. For the induction study, hepatocytes from one donor were used (50-year-old Caucasian woman). Upon thawing, the cells had viability of approximately 90% based on Trypan blue exclusion.
HTS CYP3A4 inhibition assay
An automated workstation (Biomek 2000; Beckman Coulter, Brea, CA) was used for the performance of the assay. The workstation was programmed to perform serial dilutions of the model inhibitors and for the initiation of the assay. White opaque 384-well plates (Falcon, Inc., purchased via VWR, Inc., Radnor, PA) were used. The workstation was programmed to add into each well of the 384-well plates 10 µL of hepatocytes (containing 10 000 cells) and 10 µL of hepatocyte metabolism medium containing either solvent (0.1% v/v of acetonitrile) or P450 inhibitors at the designated concentrations (at 3× of the designated concentrations). The final acetonitrile concentration was 0.033% v/v. The assay was initiated by the addition of 10 µL of 3 µM LIPA (final concentration 1 µM). The plates were returned to a cell culture incubator maintained at 37 °C, in a highly humidified atmosphere of 95% air and 5% carbon dioxide. After an incubation period of 120 min, the plates were returned to the workstation for the addition of 10 µL of luciferin detection reagent (Promega).
CYP3A4 induction assay
Cryopreserved human hepatocytes were thawed and plated onto collagen-coated 96-well plates at a cell concentration of 50 000 cells/well in 100 µL/well of Hepatocyte Plating Medium (APSciences, Columbia, MD). After culturing overnight, medium was changed to that containing 0.25 mg/mL of Matrigel (BDBioscience, Waltham, MA). After another overnight culture, medium was changed to Hepatocyte Induction Medium (APSciences) containing the designated concentrations of model inducers (100 µL/well). The hepatocytes were then cultured for 3 additional days to allow 72 h of continuous treatment without medium change. At the end of treatment, cells were washed three times with Hepatocyte Metabolism Medium followed by incubation with Hepatocyte Metabolism Medium (APSciences) containing 3 µM of LIPA. After an incubation period of 120 min, 50 µL of the medium was removed and transferred to white opaque plates followed by addition of 50 µL of luciferin detection reagent (Promega).
Data analysis
Luminescence was quantified using a multichannel plate reader (Wallac 1420; PerkinElmer, Waltham, MA). Luminescence signals were converted to pmoles of luciferin based on a standard curve generated from luciferin standard (Promega).
Enzyme specificity results are expressed as pmol luciferin generated/pmol of enzyme/min. Results for the CYP3A4 inhibition and induction assays are expressed as relative activity, which is calculated as a ratio of the activity in the presence of inhibitors or inducers to that of the solvent control using the following equation:
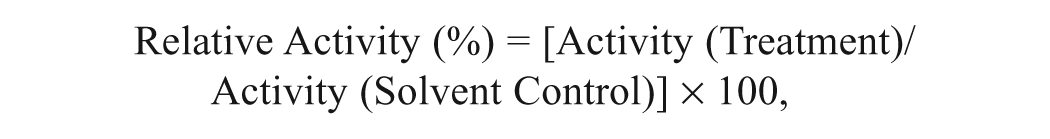
where activity represents luciferin generated in each well quantified by luminescence.
EC50 values are calculated based on nonlinear regression of a plot of relative activity versus the logarithm of inhibitor concentrations. Nonlinear regression analysis was performed using Prism software (GraphPad Software, San Diego, CA).
Statistical analysis
The statistical significance of inhibition was determined by comparing treatment to solvent control based on Dunnett’s t test using (JMP 6.0 statistical software; SAS, Cary, NC), with p ≤ 0.05 as the level of significance.
Results
Evaluation of CYP3A4 specificity of LIPA using recombinant P450 isoforms
Dose-dependent metabolism of LIPA by CYP3A4 was observed ( Fig. 1 ). When LIPA was incubated with recombinant P450 isoforms CYP3A4, CYP2C19, CYP2E1, CYP2C9, CYP1A2, and CYP2D6, significant metabolism to luciferin was observed only for CYP3A4. The results provide evidence that LIPA was a selective CYP3A4 substrate ( Fig. 1 ).
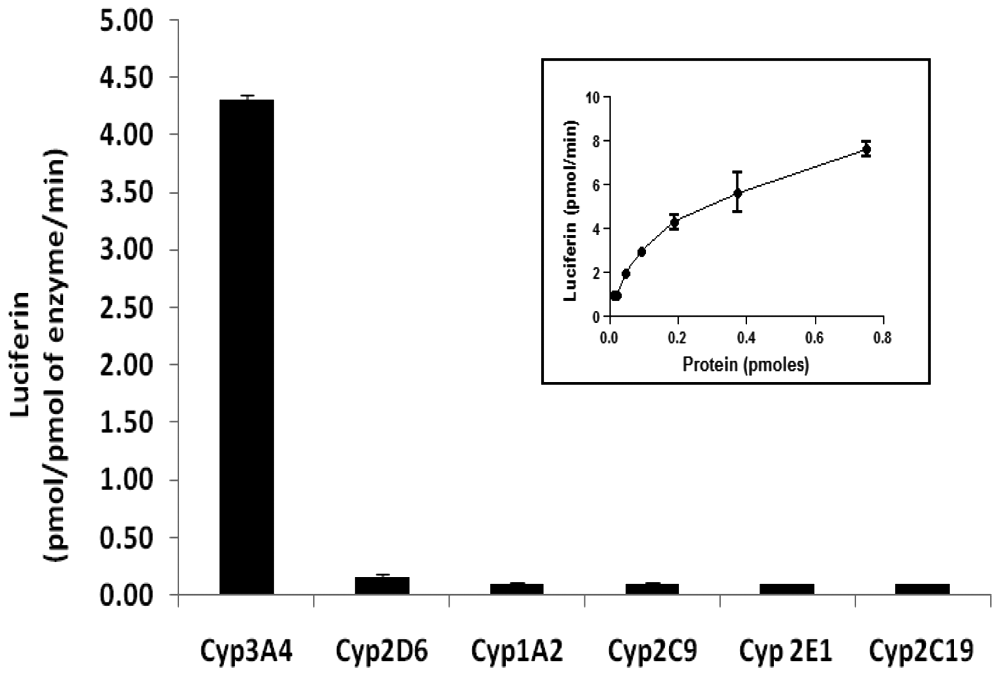
Results of in vitro incubation of luciferin IPA (LIPA; 3 µM) with six different recombinant human P450 isoforms at the enzyme concentration of 12.5 pmol/mL. Results are expressed as pmol of luciferin generated/pmol of the enzyme/min. Each error bar represents the standard deviation value of the mean of triplicate observations. Inset: Luciferin generation from LIPA by various concentrations of recombinant CYP3A4.
HTS human hepatocyte CYP3A4 inhibition assay
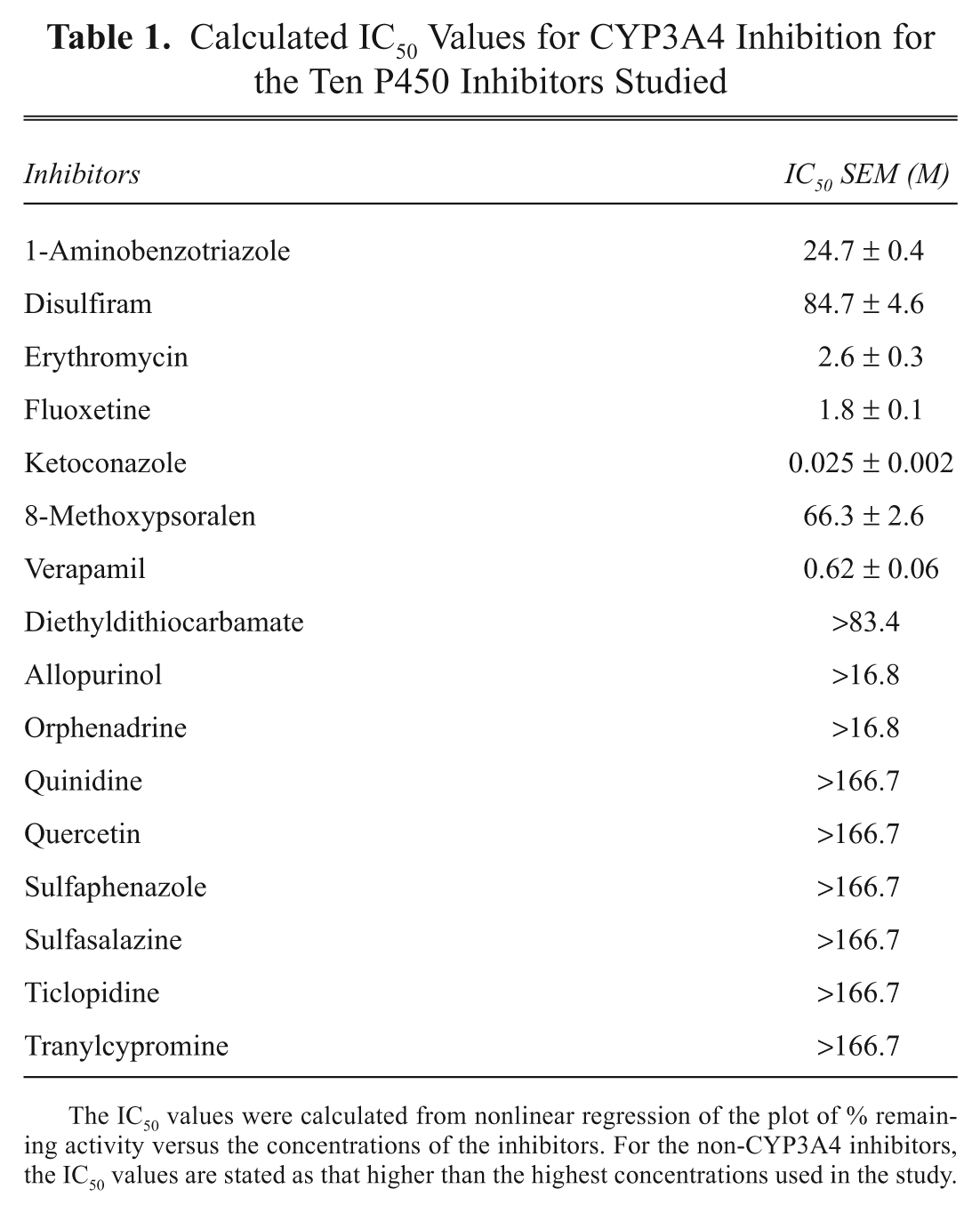
Of the ten P450 inhibitors evaluated, extensive (>80% inhibition), statistically significant, and dose-dependent inhibition was observed for chemicals that are known to inhibit CYP3A4: 1- aminobenzotriazole, erythromycin, ketoconazole, and verapamil ( Fig. 2 ). Substantially less (<50%) inhibition was observed for the chemicals that have selective inhibitory potential for non-CYP3A4 isoforms: diethyldithiocarbamate (selective for CYP2E1), quercetin (CYP2C8), quinidine (CYP2D6), sulfaphenazole (CYP2C9), ticlopidine (CYP2B6, CYP2C19, CYP2D6), and tranylcypromine (CYP2A6) ( Fig. 3 ). The IC50 values calculated for CYP3A4 inhibition in the assay are shown in Table 1 .
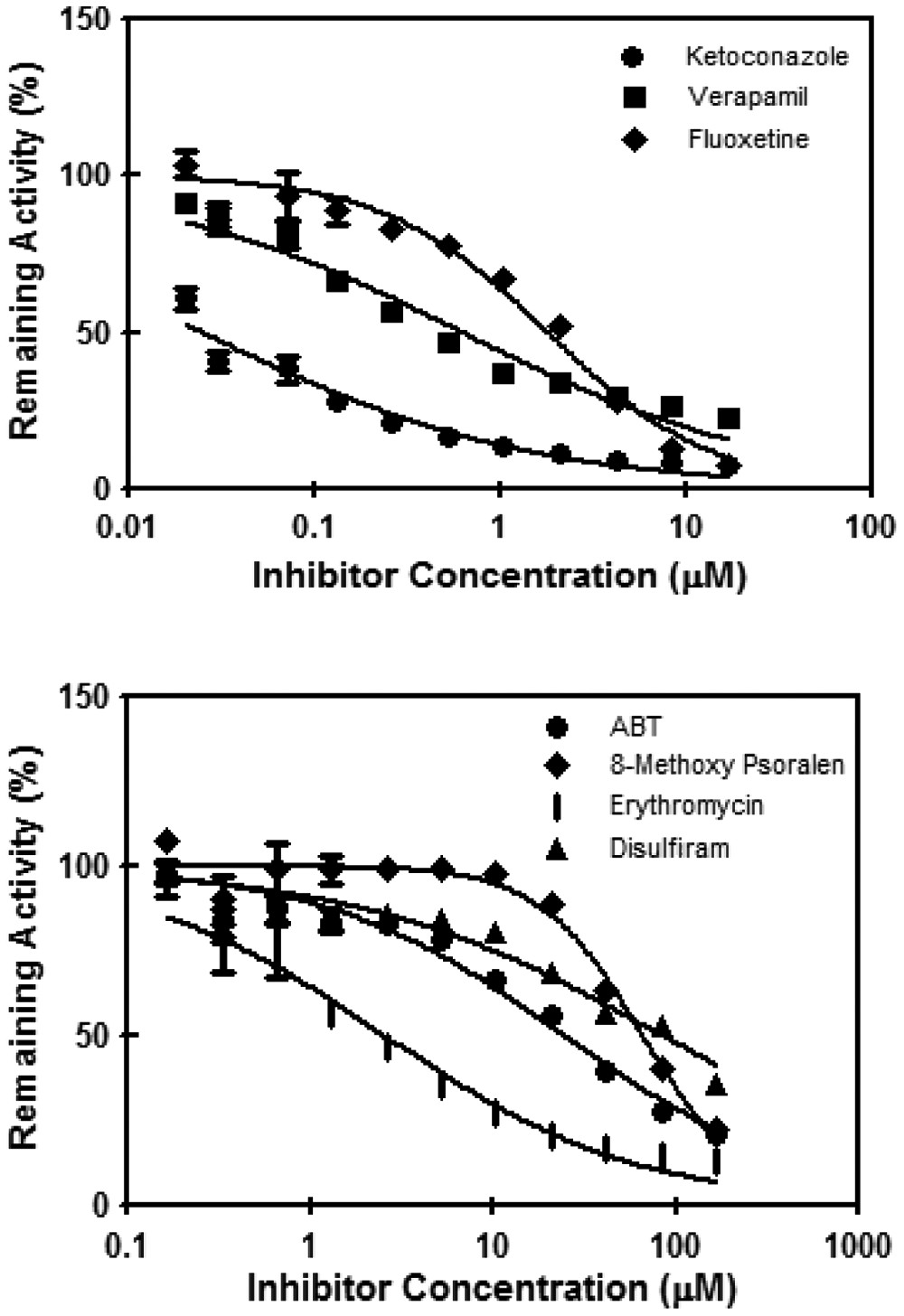
Results of CYP3A4 inhibitors in the high-throughput screening human hepatocytes CYP3A4 inhibition assay. The relative activity, expressed as percent of the solvent control activity, was plotted against the logarithm of inhibitor concentrations. Each error bar represents the standard deviation value of the mean of quadruplicate observations. Some error bars are not readily discernable because of the relatively small values versus the size of the symbols. ABT, 1-aminobenzotriazole.
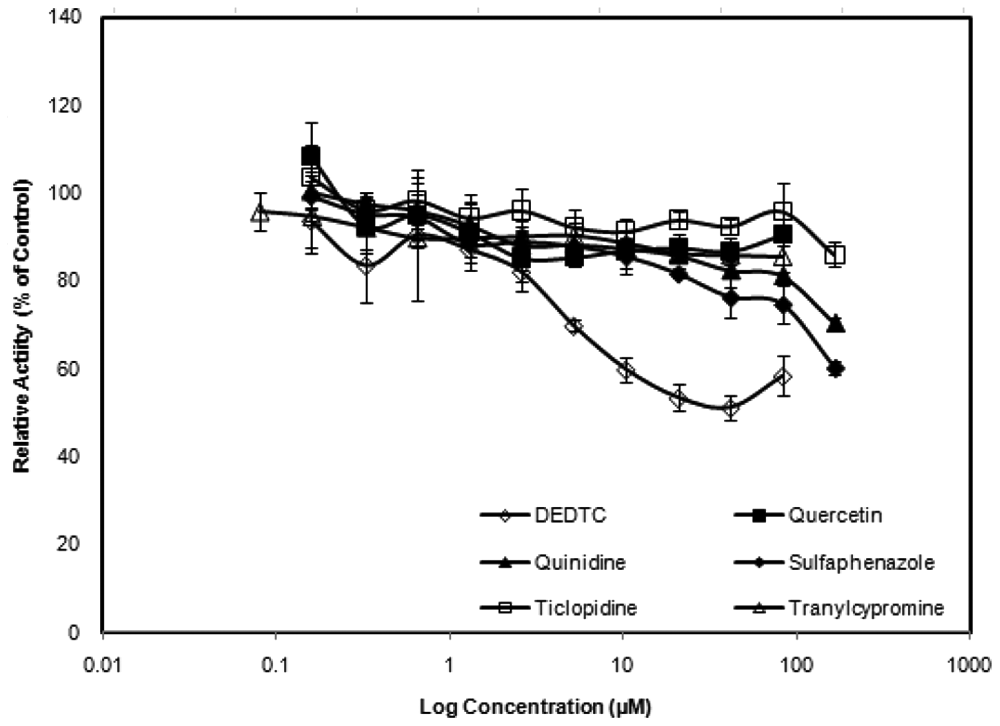
Results of the non-CYP3A4 inhibitors diethyldithiocarbamate (DEDTC), quercetin, quinidine, sulfaphenazole, ticlopidine, and tranylcypromine in the high-throughput screening human hepatocytes CYP3A4 inhibition assay. The relative activity, expressed as percent of the solvent control activity, was plotted against the logarithm of inhibitor concentrations. Each error bar represents the standard deviation value of the mean of quadruplicate observations. Some error bars are not readily discernable because of the relatively small values versus the size of the symbols.
Calculated IC50 Values for CYP3A4 Inhibition for the Ten P450 Inhibitors Studied
The IC50 values were calculated from nonlinear regression of the plot of % remaining activity versus the concentrations of the inhibitors. For the non-CYP3A4 inhibitors, the IC50 values are stated as that higher than the highest concentrations used in the study.
HTS human hepatocyte CYP3A4 induction assay
The known CYP3A4 inducers rifampin, phenobarbital, carbamazepine, phenytoin, troglitazone, rosiglitazone, and pioglitazone yielded extensive, dose-dependent induction of LIPA metabolism ( Figs. 4 and 5 ), whereas the CYP1A2 inducers omeprazole and 3-methylcholanthrene did not display any induction in the LIPA metabolism (data not shown).
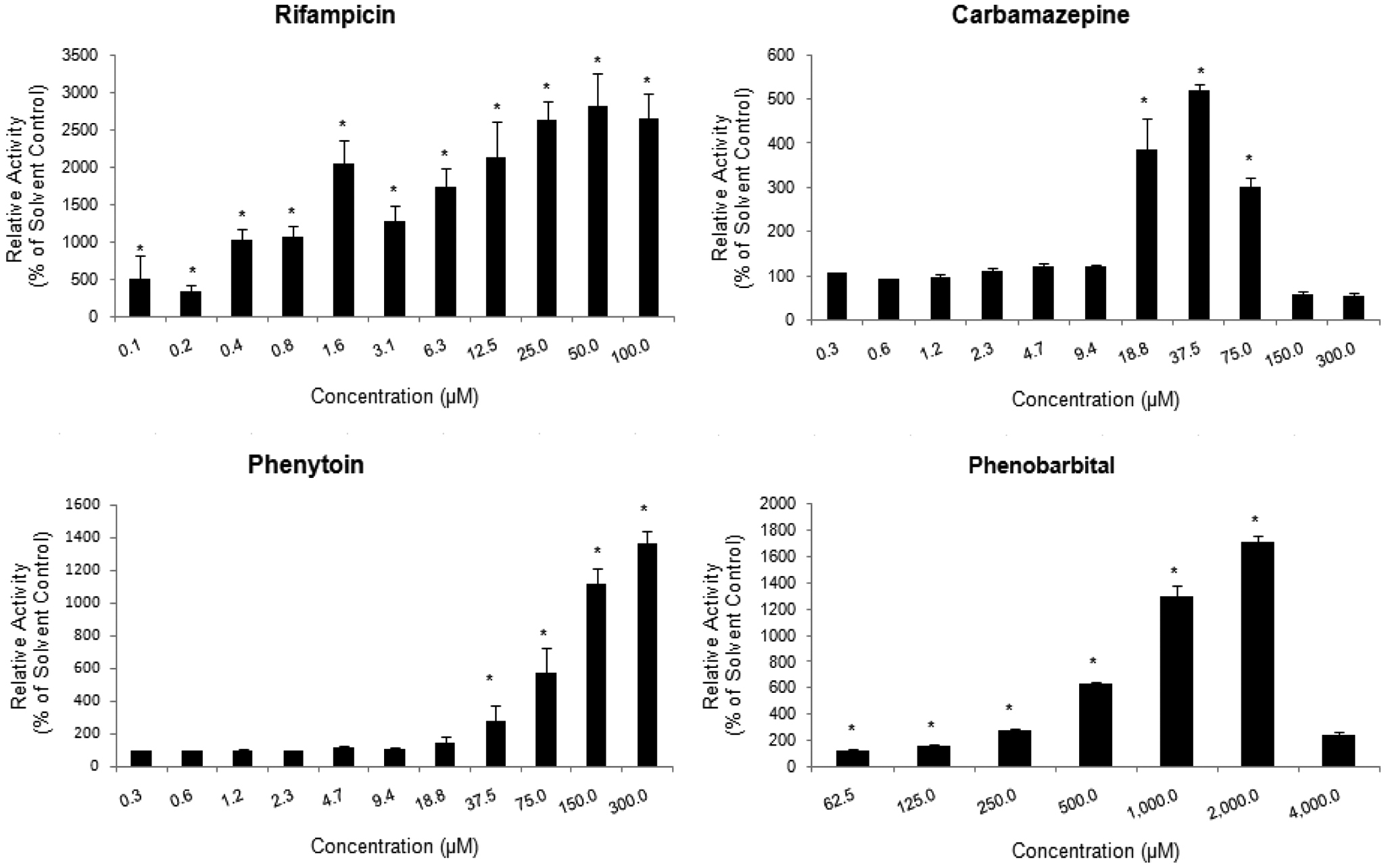
Effects of CYP3A4 prototypical inducers carbamazepine, rifampin, phenytoin, and phenobarbital on luciferin IPA (LIPA) metabolism in human hepatocytes. The relative activity, expressed as percent of the solvent control activity ± SD, was plotted against inducer concentration. * represents statistically significant (p ≤ 0.05) induction as compared to solvent control.
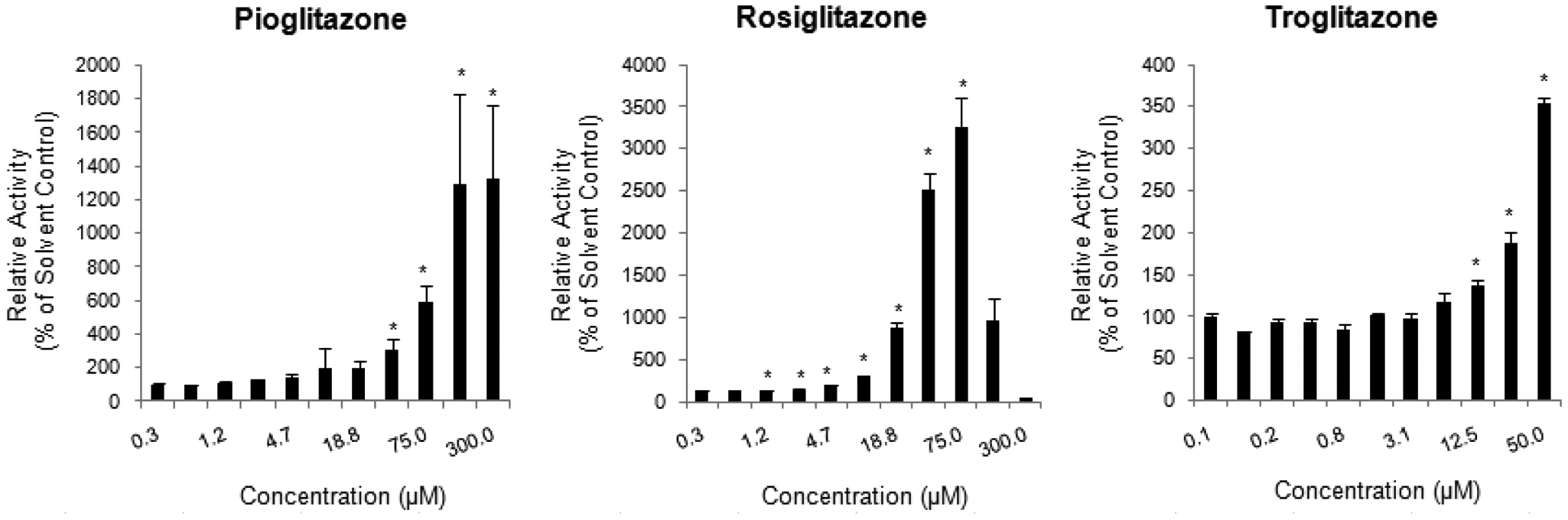
Results of three thiazolidinedione structural analogs pioglitazone, rosiglitazone, and troglitazone in the high-throughput screening human hepatocyte CYP3A4 induction assay. The relative activity, expressed as percent of the solvent control activity ± SD, was plotted against inducer concentration. * represents statistically significant (p ≤ 0.05) induction as compared to solvent control.
Discussion
Pharmacokinetic drug–drug interactions, especially CYP3A4-based events, are known to have serious, sometimes fatal, outcomes in patients. Early elimination of chemical structures with DDI liabilities would greatly facilitate the drug discovery process. The advantage of using LIPA to quantify CYP3A4 activity is that its metabolism can be quantified using a plate reader that is fast, is relatively inexpensive, and, most important, eliminates the need for LC/MS, which is often not readily assessable to drug discovery scientists because of the lack of expertise and availability to the instrument.
The specificity of LIPA for CYP3A4 is illustrated by three independent lines of evidence: (1) Recombinant P450 isoforms showed that CYP3A4 was the only isoform that could metabolize LIPA to luciferin. (2) In the hepatocyte CYP3A4 inhibition assay, concentration-dependent, statistically significant, and extensive inhibition (maximum inhibition of >80%) was observed with the known CYP3A4 inhibitors (1-aminobenzotriazole, erythromycin, ketoconazole, and verapamil), and substantially lower levels of inhibition (maximum inhibition of <50%) were observed with the known non-CYP3A4 inhibitors (diethyldithiocarbamate, quercetin, quinidine, sulfaphenazole, ticlopidine, and tranylcypromine). Of the non-CYP3A4 inhibitors, diethyldithiocarbamate had the highest inhibitory effect (maximum inhibition of near 50%), a result similar to that reported by Eagling et al. 22 using human liver microsomes and quantifying CYP3A4 activity via testosterone 6b-hydroxylation. (3) In the hepatocyte CYP3A4 induction assay, the known CYP3A4 inducers rifampin, phenobarbital, carbamazepine, phenytoin, and the thiazolidinediones troglitazone, rosiglitazone, and pioglitazone demonstrated dose-dependent induction, whereas no induction in LIPA metabolism was observed with the CYP1A inducers omeprazole and 3-methylcholanthrene.
The 384-well hepatocyte CYP3A4 inhibition assay is an extension of our previous studies, which introduced the use of human hepatocytes in P450 inhibition studies.15,20,23 Although the evaluation of P450 inhibition is traditionally performed with liver microsomes and recombinant CYP enzymes,24,25 intact hepatocytes may provide useful information to improve the accuracy of the prediction of in vivo effects. A chemical, for instance, may be metabolized by non-CYP pathways to a metabolite that is a potent P450 inhibitor and therefore would be inhibitory in hepatocytes but not in microsomes or recombinant CYP enzymes. Gemfibrozil, for instance, requires glucuronidation for its CYP2C8 inhibitory effects and is a potent CYP2C8 inhibitor in hepatocytes but less so in liver microsomes or recombinant CYP2C8. 26 The presence of active transporters in human hepatocytes, including cryopreserved hepatocytes, also suggests that an inhibitor may be actively accumulated inside the cells, leading to a substantially higher concentration and a correspondingly higher inhibitory effect that would not be observed using cell-free systems. 27 In the HTS human hepatocyte CYP3A4 inhibition assay described here, 384-well plates were used to reduce the quantity of hepatocytes, reagents, and the chemical to be evaluated. The use of LIPA as a CYP3A4 substrate substantially enhances the efficiency of the assay, eliminating analytical time that traditionally is considered the “bottleneck” for such assays. The use of robotics allowed rapid and accurate delivery of relatively small volumes of reagents into the 384-well plates. The accuracy of the assay is demonstrated by the relatively low coefficient of variation (standard deviations <10% of mean values) of the results. Most important, the results with the model P450 inhibitors demonstrate that this human hepatocyte HTS assay can readily distinguish CYP3A4 inhibitors from noninhibitors.
Human hepatocyte cultures have been universally accepted as the “gold standard” for the evaluation of P450 induction.7,28,29 We have made several improvements to enhance the efficiency of the induction assay. First, the use of 96-well plate format allows one to evaluate four chemicals in seven concentrations in triplicate in one single plate, which is a significant improvement over the traditional assay using 24-well plates. Second, the elimination of daily medium change during the treatment period greatly decreased the time and effort required in this assay. As described earlier, the assay readily identified the known CYP3A4 inducers from the non-CYP3A4 inducers, suggesting that it can be used routinely for the evaluation of CYP3A4 induction potential of new chemical entities.
It is now known that the lack of efficacy and the presence of adverse drug effects are the major reasons for clinical failure of new drug candidates. It is therefore advantageous for drug discovery scientists to routinely evaluate adverse drug effects in addition to pharmacological effects to allow the logical selection of the drug candidates with the highest potential of success for further development. The hepatocyte-based assays for CYP3A4 inhibition and induction described here are intended to be used by drug discovery scientists so that these critical adverse drug properties can be evaluated hand-in-hand with pharmacology and in vitro cytotoxicity assays, allowing the selection of the new chemical entities with the highest potential for success for further development.