
Abstract
Antibody-based therapeutics play a vital role in the treatment of certain cancers; however, despite commercial success, various strategies are being pursued to increase their potency and hence improve patient outcomes. The use of antibodies to deliver a cytotoxic payload offers a promising alternative for more efficacious therapies. Immunotoxins are composed of an internalizing antibody fragment linked to a bacterial or plant toxin. Once internalized, the payload, such as Pseudomonas exotoxin A (PE), blocks protein synthesis and induces apoptosis. Typically, immunotoxins are developed by first isolating a tumor-specific antibody, which is then either chemically linked to a toxin or reengineered as a fusion protein. Here, the authors describe the development of Fusogenics, an immunotoxin-based screening method that selects internalizing tumor-specific antibodies using a functional assay. Selected immune library clones were characterized and shown to be selective against normal tissues and specific to tumor tissues. In summary, the Fusogenics immunotoxin platform represents a unique, single-step selection approach combining specificity and functionality to isolate novel internalizing tumor-specific antibody fragments with potential for direct clinical application in the treatment of cancer.
Introduction
I
Despite the commercial success of therapeutic Abs, approaches to further improve potency such as mutation of Fc residue(s) or de-fucosylation of the carbohydrate chains are currently being explored to improve clinical benefit. 10 Another approach is to develop antibody drug conjugates (ADCs) or immunotoxins that use the antibody as a vehicle to specifically deliver a cytotoxic payload such as a small molecule or a protein toxin to cancer cells. 11,12 ADCs rely on linker technology to chemically conjugate the payload to the antibody, whereas recombinant immunotoxins are fusion proteins that contain a cytotoxic protein genetically linked to an antibody fragment. To be efficacious, an immunotoxin fusion protein engineered with a scFv or Fab fragment requires internalization without receptor cross-linking. Once internalized, bacterial toxins such as Pseudomonas exotoxin A (PE) and plant toxins such as bouganin, gelonin, or saporin prevent protein synthesis leading to cell death. 12 As a consequence of their cytotoxicity to mammalian cells, immunotoxins are generally expressed in microbial cells and purified either from inclusion bodies or the periplasmic space. The clinical benefits of immunotoxins have been demonstrated mostly with hematological cancers and in the case of solid tumors using local delivery strategies. 13,14 Thus, given the need to select more potent cancer therapeutic drugs, a possible strategy is to develop a screening approach using an immunotoxin format that has the potential to simultaneously identify tumor-targeting antibodies and rapidly assess their cytotoxicity in a single step without the need for molecular reengineering.
In this study, we investigated the immunotoxin concept in a soluble screening format to select and identify novel internalizing Ab candidates from immune libraries. As a proof of concept, an internalizing anti-EpCAM Fab-PE recombinant fusion protein was used to develop a functional screening assay for measuring the level of PE-induced apoptosis. Subsequently, the concept was extended to a screening platform—namely, Fusogenics—which selects internalizing, tumor-specific Ab fragments from a Fab-PE immune library created from the plasma B cell population of colon cancer patients. The biological characterization, tumor reactivity, and cytotoxic potential of 1 clone—namely, VB6-314-PE—are used to demonstrate the potential of this novel screening approach.
Materials and Methods
Cell culture
All tumor cell lines were purchased from ATCC (Manassas, VA) with the exception of CAL-27, which was obtained from DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany). Cell lines were cultured in a humidified incubator at 37°C in presence of 5% carbon dioxide and maintained in their respective medium as per the provider’s recommendations.
Fusogenics immune library construction
Lymphocytes, sourced from lymph nodes of colon cancer patients (Cureline, San Francisco, CA), were suspended in 1 mL Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and plasma B cells negatively selected in the presence of 250 ng and 50 ng of antihuman IgD (BD Biosciences, Mississauga, Ontario, Canada) and antihuman Thy-1 (Serotec, Raleigh, NC) biotinylated antibodies, respectively. After 90 min on ice, cells were centrifuged and resuspended in 1 mL DMEM, 10% FBS. An equal volume of magnetic beads coated with streptavidin was added to the cells, and after 1 h on ice, naive B cells and T cells were separated magnetically and the media containing enriched plasma B cells collected.
mRNAs from enriched plasma B cells were extracted using the Oligotex™ kit (Invitrogen, Toronto, Ontario, Canada) and the first-strand cDNA synthesized using the SuperScriptIII reverse transcriptase kit (Invitrogen). Library clones were expressed in Escherichia coli supernatant as a Fab recombinant fusion protein comprising an antibody Fd fragment genetically linked to a truncated form of Pseudomonas exotoxin A, PE252-608. Both chains, preceded by a PelB leader sequence, were cloned into the pING3302 plasmid (Xoma, Berkeley, CA) as a dicistronic unit under the control of an arabinose promoter. The VL kappa repertoire was amplified by PCR with a mixture of 5′ primers corresponding to the different subclasses and containing the SfiI restriction site, a 6xHis tag for purification, and a 3′ end constant kappa chain primer containing a XhoI restriction site. The PCR product was digested with SfiI and XhoI restriction enzymes and ligated into the EcoRI-ApaI-CH-PE-PelB-SfiI-XhoI/pING3302 plasmid ( Fig. 1A ). EasyShock™ 10B electrocompetent cells (BioRad, Mississauga, Ontario, Canada) were then electroporated with the purified ligation reaction (Zymo Research, Burlington, Ontario, Canada). The VH gamma repertoire was obtained with a mixture of 5′ primers and a 3′ constant gamma chain domain primer containing the NcoI and ApaI restriction site, respectively. The VH PCR product was linked to the EcoRI-PelB leader sequence into an intermediate plasmid using the NcoI and ApaI restriction sites. The full insert was digested with EcoRI and ApaI restriction enzymes and randomly cloned into CH-PE-PelB-VL-CL/pING3302 plasmids ( Fig. 1B ). The ligation reaction and transformation in 10B cells was performed as previously described. The final recombinant plasmids containing randomly paired variable domains were then electroporated into JM109 and 10F E. coli strains (Zymo Research; Invitrogen).
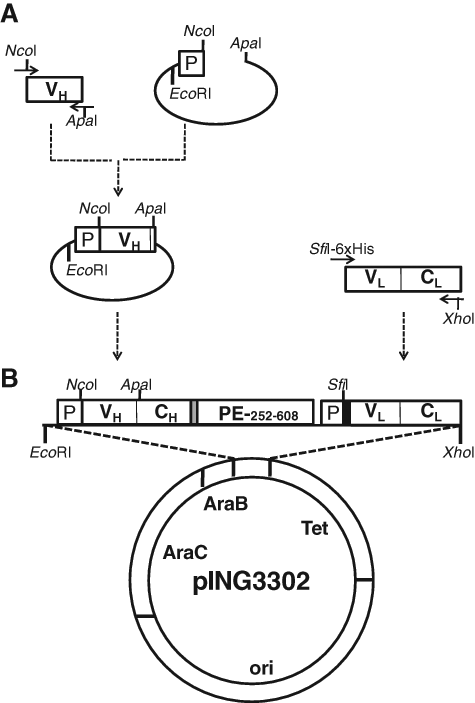
Schematic representation of Fusogenics immune library construction. VH, VL, CH, CL, PE, and P abbreviations correspond to the variable heavy and light chain and heavy and kappa chain conserved domains and truncated form of Pseudomonas exotoxin A from amino acids 252 to 608, to the PelB leader sequence, respectively. (
To create the anti-EpCAM Fab-PE fusion protein—namely, VB6-845-PE—the 5′ NcoI–3′ ApaI restriction sites of the variable heavy chain 4D5MOCB were introduced by PCR reaction using oligonucleotide primers. 15 The conserved kappa constant domain and the 5′ SfiI–3′ XhoI restriction sites were added to the 4D5MOCB variable light chain by splice overlapping extension PCR.
Fusogenics immune library screening
Transformed E. coli JM109 cells were plated onto LB-agar Q-tray plates (22 × 22cm) supplemented with 25 µg/mL of tetracycline. To maximize the efficiency of the screening procedure, single colonies were picked with a colony picker (Genetix, New Milton, Hampshire, UK) and all liquid transfers performed with a BiomekFX (Beckman Coulter, Fullerton, CA). Briefly, 96-well plates containing 150 µL of 2xYT supplemented with 25 µg/mL of tetracycline were inoculated with a single colony using automated picking and incubated overnight at 37°C in a humidified shaking incubator (Liconic Instruments, Woburn, MA). Overnight incubation allowed all clones to reach the growth phase plateau, thereby standardizing the inoculums used for the induction plate. A 20-µL culture from each well was transferred into a 96-well daughter induction plate containing 130 µL of terrific broth (TB) and incubated at 37°C with constant shaking. Then, 20 µL of 50% sterile glycerol was added to each well of the 2xYT plates, which were then stored at −20°C. After 7 h of incubation, the TB plates were induced with 0.2% L-arabinose and incubated overnight at 25°C. After centrifugation at 5000 rpm for 30 min, 10 µL of supernatant was added to 96-well FMAT plates preseeded with SW-480 colon tumor cells at a density of 8000 cells per well (in DMEM supplemented with 1% penicillin-streptomycin) and incubated overnight in a 5% CO2 incubator set at 37°C. The remaining supernatant was stored at −20°C in 96-well plates for further analysis. Using the 8200 Cellular Detection System (Applied Biosystems, Streetville, Ontario, Canada), the percentage of apoptotic cells was measured by the addition of 20 µL of Annexin V buffer (140 mM NaCl, 12.5 nM CaCl2, and 10 mM HEPES) containing Annexin V coupled to Alexa-647 (Invitrogen) diluted 1/3000 and 0.75 nM CentriRed (Applied Biosystems) for 50 min at room temperature (RT). E. coli JM109 cells transformed with either VB6-845-PE or the pING3302 plasmid alone were used as positive and negative controls, respectively. Wells showing a level of apoptosis of at least 2-fold higher than the negative control were retested in 3 independent wells to demonstrate reproducibility. To finalize the selection, candidate clones were then retested in triplicate against SW-480 colon tumor cells and the B cell lymphoma, CA46.
Dot-blot and enzyme-linked immunosorbent assay quantification
To determine the percentage of expressing immune library clones, 50 µL of induced supernatant from randomly chosen immune library clones was transferred onto a nitrocellulose membrane using a mini-fold apparatus. After blocking with a 1× Tris-buffered saline (TBS) 3% bovine serum albumin (BSA) solution for 1 h, the membrane was incubated with an antihuman kappa horseradish peroxidase (HRP) conjugate (Sigma-Aldrich, St. Louis, MO) (1/1000) for 2 h and bound antibody detected using DAB solution (Pierce, Rockland, IL).
To quantify the level of expression of immune library clones, a 96-well plate (Immulon 1B) was coated with 10 µg/mL of rabbit anti–Pseudomonas exotoxin A (Sigma-Aldrich) overnight at 4°C and blocked with 1% BSA for 1 h at 22°C. Induced supernatants diluted from 1/320 to 1/2560 were added to the plate and incubated for 2 h at 22°C. To detect bound Fab-PE fusion protein, an antihuman kappa light chain HRP conjugate (Sigma-Aldrich) was added to each well and, after 1 h at 22°C, the reaction developed with 3,3′,5,5′-tetramethylbenzidine. A standard curve was obtained with purified VB6-845-PE and induced pING3302 supernatant used as a negative control.
Expression and purification of immune library clones
Fermentation of immune library clones was performed in a 15-L CHEMAP fermenter using TB medium. Cultures were induced at an OD600 of 2 with a mixture of feed (50% glycerol) and inducer (200 g L-arabinose). At 24 h postinduction, the culture was harvested, centrifuged at 8000 rpm for 30 min, concentrated, and diafiltered against 20 mM sodium phosphate (pH 7.0). The supernatant was then applied to a charged chelating sepharose column and the column washed with 10 mM imidazole. Bound Fab-PE was eluted with 250 mM imidazole. To obtain purity >90%, the eluate of the chelating sepharose column was applied onto a size exclusion column S200 equilibrated with 20 mM sodium phosphate, 150 mM NaCl, pH 7.5. Purity following the size exclusion column was confirmed by colloidal blue staining and identity by Western blotting using rabbit anti–Pseudomonas exotoxin A (Sigma-Aldrich) followed by goat antirabbit HRP conjugate (The Binding Site, San Diego, CA).
Sequence analysis
Nucleotide sequence of the heavy and light chain variable domains was performed using the GenomeLab™ Dye Terminator Cycle sequencing (Beckman Coulter, Mississauga, Ontario, Canada) with specific primers. Sequences were analyzed using NCBI IgBlast to determine CDR regions and V gene usage.
Clone characterization and selection
Cell surface reactivity and internalization
The reactivity of immune library clones was determined against a panel of tumor cells representative of cancer indications by flow cytometry using a FACS Calibur (BD Biosciences). Briefly, tumor cells (1 × 106/mL) were incubated with 10 µg/mL of purified Fab-PE for 2 h on ice. After washing away the unbound material, bound Fab-PE was detected using a biotinylated goat antihuman H&L chain antibody (Pierce) followed by Cy-5-streptavidin (Pierce). Cells were analyzed on a FACS Calibur following propidium iodide (Molecular Probes, Eugene, OR) staining. The rate of internalization was determined by measuring the Fab-PE protein bound to SW-480 tumor cells after incubation at 37°C for 15, 30, 60, and 120 min. The results were presented as percent decrease in median fluorescence (MF) versus a 4°C sample (100%).
Affinity
The functional affinity of individual clones was determined by flow cytometry. 16 Briefly, MDA-MB-435S cells (6 × 105/mL) were incubated with increasing concentrations of Fab-PE for 2 h at 4°C to establish a saturation curve. Bound Fab-PE was detected as described above. The functional affinity, expressed as the dissociation constant KD, was calculated by the Lineweaver-Burk method of plotting the inverse of the MF as a function of the inverse of the antibody concentration. The dissociation constant was determined by the following equation: 1/F = 1/Fmax + (KD/Fmax)(1/[Ab]), where F corresponds to the background subtracted MF, and Fmax was calculated from the plot.
Potency
The potency was measured by an MTS assay (Promega, Madison, WI). Briefly, SW-480 and CA46 cells were seeded at 5000 cells per well and incubated at 37°C for 3 h. Purified Fab-PE was added to the cells over a range of concentrations, incubated for 3 days, and an IC50 interpolated from the resulting plot.
Tumor and normal tissue reactivity
The selectivity and specificity of Fab-PE fusion proteins was assessed against critical normal and tumor tissues using immunohistochemistry. Briefly, formalin-fixed tissues slides (TriStar, Rockville, MD) were deparaffinized in xylene, hydrated in alcohols, and blocked for endogenous peroxidase with 3% hydrogen peroxidase. For antigen retrieval, slides were heated to 125°C in target retrieval solution (Dako Diagnostics, Mississauga, ON) in a decloaking chamber. Fab-PE was then added to the tissues and incubated for 1 h at RT. After washing, bound Fab-PE was detected with rabbit anti–Pseudomonas exotoxin A (Sigma-Aldrich) followed by EnVision-HRP (Dako Diagnostic) and cytomation liquid DAB plus substrate chromogen system (Dako Diagnostic). Slides were counterstained in Harris hematoxylin (Fisher, Ottawa, Ontario, Canada), coverslipped using cytomation mounting media (Dako Diagnostic), and analyzed for cell surface staining by a board-certified pathologist.
Results
Fusogenics: screening parameters
The feasibility of this approach, and in particular the expression level in E. coli supernatant and screening parameters, was tested using VB6-845-PE, an internalizing anti-EpCAM Fab fragment genetically linked to PE252-608. To select the optimal electro-competent E. coli strains for supernatant secretion, 10F and JM109 cells were transformed with the recombinant plasmid. The level of expression was assayed in either a 96-well plate or 96-well deep plate and compared to those typically obtained under shake-flask conditions. Western blot analysis showed that VB6-845-PE expression level in induced E. coli supernatants was similar with either type of 96-well plate but, as expected, lower when compared to shake-flask levels ( Fig. 2A ). With respect to host strain expression, VB6-845-PE expression was higher in JM109 supernatant (~2.5 µg/mL by enzyme-linked immunosorbent assay [ELISA]) as compared to 10F, thus making JM109 the preferred host cell.
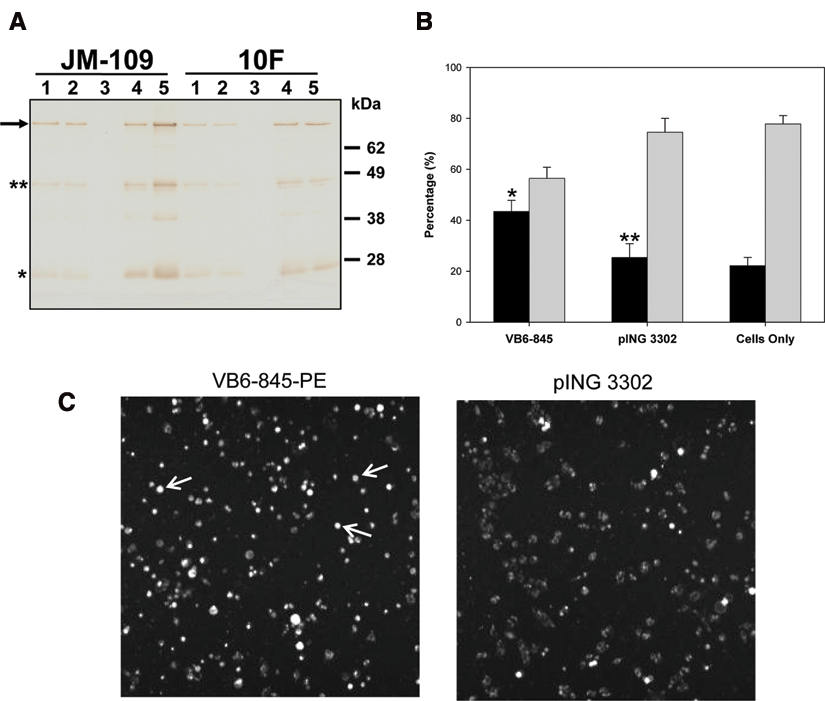
Expression and cytotoxicity of soluble VB6-845-PE expressed in Escherichia coli. (
Inhibition of E. coli growth during the functional assay was a prerequisite to ensure a readable outcome. Therefore, preliminary experiments performed in a 96-well plate showed that a mixture of 10 to 30 µL of JM109-induced supernatants with 100 µL of DMEM containing antibiotics was E. coli free for up to 24 h at 37°C. Therefore, the cytotoxicity of 10, 20, and 30 µL of VB6-845-PE and pING3302 JM109-induced supernatants was measured using the EpCAM-positive tumor cell line SW-480 and an EpCAM-negative B cell lymphoma, CA46, seeded on a FMAT plate (
Fusogenics: immune library creation
To enrich the immune library with tumor-reactive antibody fragments, only plasma B cells isolated from the lymph nodes of 3 colon cancer patients were used as starting material. After mRNA extraction from 2.6 × 106 plasma B cells, only IgG-derived VH fragment and VL kappa repertoires were amplified by using an antisense oligonucleotide primer homologous to the 5′ end constant domain. A library representing a repertoire of Fab genes was then assembled as a discitronic unit using a VH and VL cloning cassette. The size of the library, generated from 4 independent electroporations using a single ligation reaction, was estimated at 500,000 unique clones. The dot-blot analysis of random colonies grown and induced in a 96-well plate showed that approximately 70% of the clones produced a soluble Fab-PE fusion protein in the supernatant (data not shown). A third of the clones had an expression level estimated at approximately 2.5 µg/mL, another third ~1 µg/mL, and the remaining below 0.5 µg/mL, but they were all within the functional range of the assay. The diversity of the library was evaluated by analyzing the sequences of 30 expressing clones. The sequence analysis revealed 30 unique sequences, and the CDR3 length of the variable heavy chains showed a distribution between 10 and 21 amino acid residues ( Fig. 3A ). Different heavy and light gene families were represented in the library; however, more than 65% of the clones had either VH3 or VK1 subclasses ( Fig. 3B ). As well, alignment of the variable domains with germinal sequences revealed that 88% of the heavy and light chains contained on average 5.5% and 4% mutated residues, respectively, corresponding to somatic mutations. As expected, the number of mutations in the CDR1 and CDR2 loops was greater by 2- to 3-fold compared to framework ( Fig. 3C ).
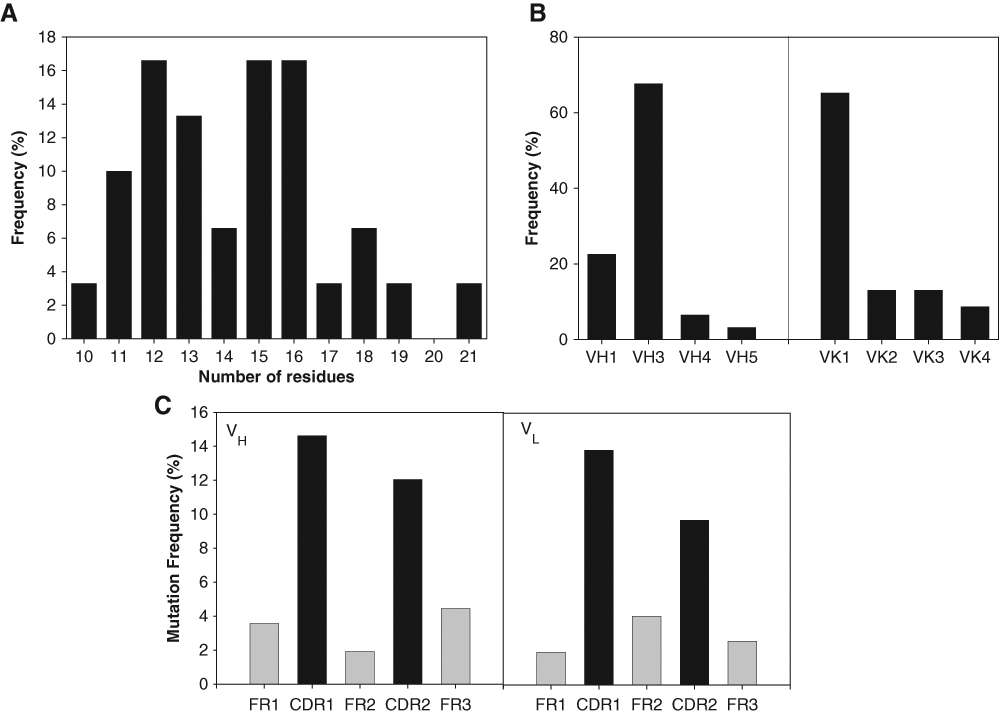
Sequence analysis of immune library clones. (
Immune library screening
The immune library was screened against SW-480 tumor cells that had been detached using PBS-EDTA, to avoid proteolytic degradation of potentially interesting cell surface antigens. Following the second round of positive selection, ~1.7% of the clones had an apoptotic level that was at least 2-fold higher than the pING3302 negative control. Of these, only 4.2% were cytotoxic to SW-480 but not CA46, suggesting specificity toward epithelial markers and resulting in an overall success rate of 0.07% for the screening procedure. To prioritize selected clones, binding to SW-480 cells was measured by flow cytometry using induced supernatant (data not shown). Clones with the highest binding activity were purified and tested against SW-480 and CA46 cell lines. As an illustration, VB6-314-PE clone is shown (
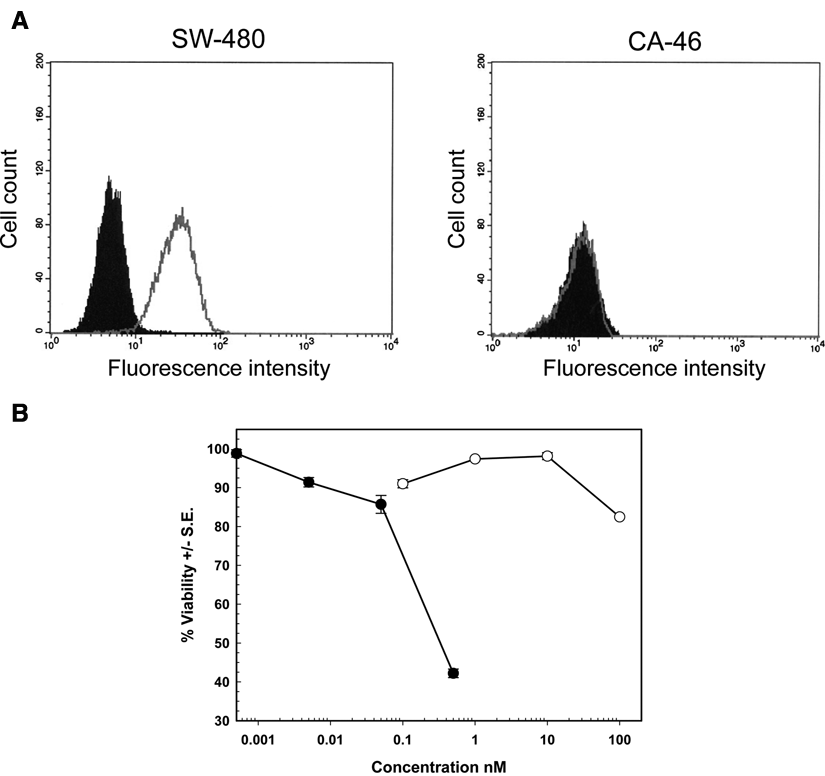
Biological activity of a purified, representative clone from the immune library. VB6-314-PE at 10 µg/mL was mixed with SW-480 or CA46 cells for 2 h at 4°C. Bound VB6-314-PE was measured by flow cytometry, and the filled area corresponds to the negative control (secondary antibody alone) and the gray line to VB6-314-PE specific binding. (
Biological characterization of selected clones
To assess the selectivity of prospective clones for tumor versus normal, the antibody fragment was tested on a low-density (LD) array of critical normal tissues by immunohistochemistry. If minimum to no cell surface staining was observed, then the antibody fragment was tested in a high-density (HD) formalin-fixed tumor microarray. Subsequently, a panel of tumor cell lines was tested by flow cytometry to confirm that the cognate antigen is also present on established cell lines. As illustrated with VB6-314-PE, only minimal cell surface staining on critical normal tissues was observed with lung, colon, and stomach ( Table 1 ). However, in these instances, the maximum score for membrane staining is only 1+ with no more than 20% of cells showing positive staining. In contrast, VB6-314-PE reactivity was detected on all tumor types tested but in particular kidney, head and neck, and prostate tissues ( Table 2 ). The intensity and percent tumor membrane staining increased compared to critical normal, suggesting a marked overexpression of the antigen. For most tumor tissues, the 2+ staining also corresponded to an increased percentage of membrane-stained cells. It is noteworthy that, although lung tumor tissue exhibited a greater percentage of cells with more intense membranous staining, the frequency of positive cores was less than observed in the normal lung panel. The higher frequency of normal lung tissue cores being stained may be explained by the unique architecture of lung tissue. The porous appearance of normal lung tissue makes the core susceptible to nonspecific staining commonly seen along tissue edges. Conversely, the cellular aggregation of the epithelium in lung tumor tissue precludes an edge effect; hence, fewer samples show positive staining.
Critical Normal Tissue Microarray
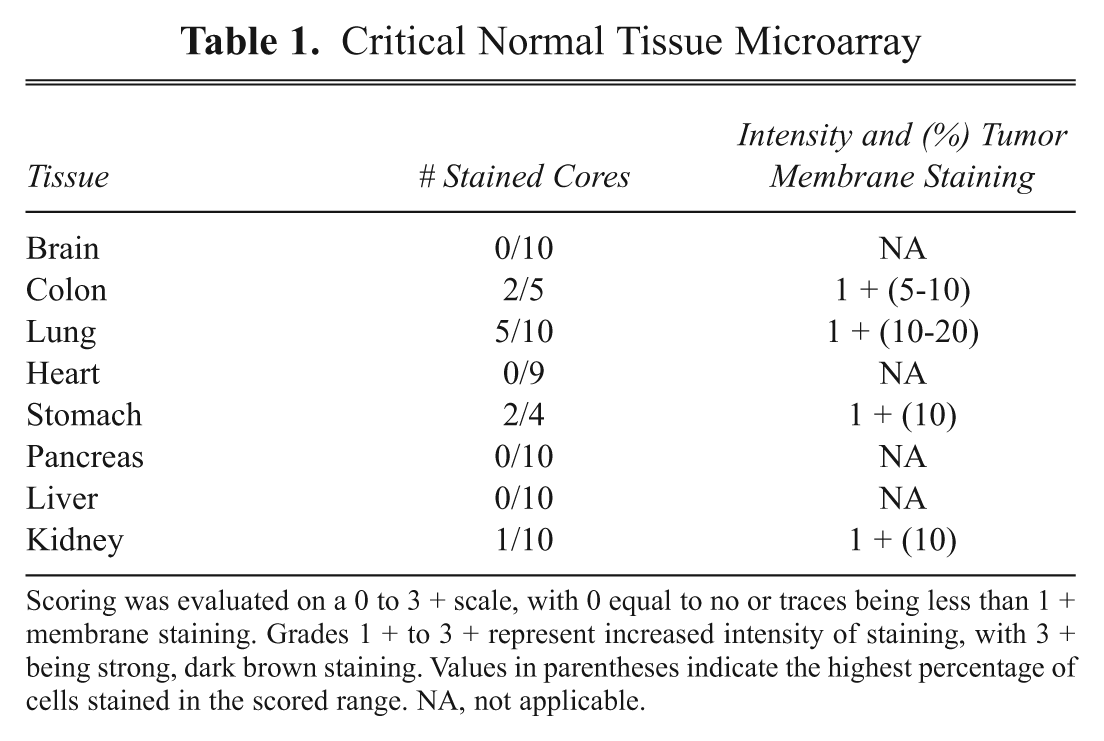
Scoring was evaluated on a 0 to 3 + scale, with 0 equal to no or traces being less than 1 + membrane staining. Grades 1 + to 3 + represent increased intensity of staining, with 3 + being strong, dark brown staining. Values in parentheses indicate the highest percentage of cells stained in the scored range. NA, not applicable.
Tumor Tissue Microarray
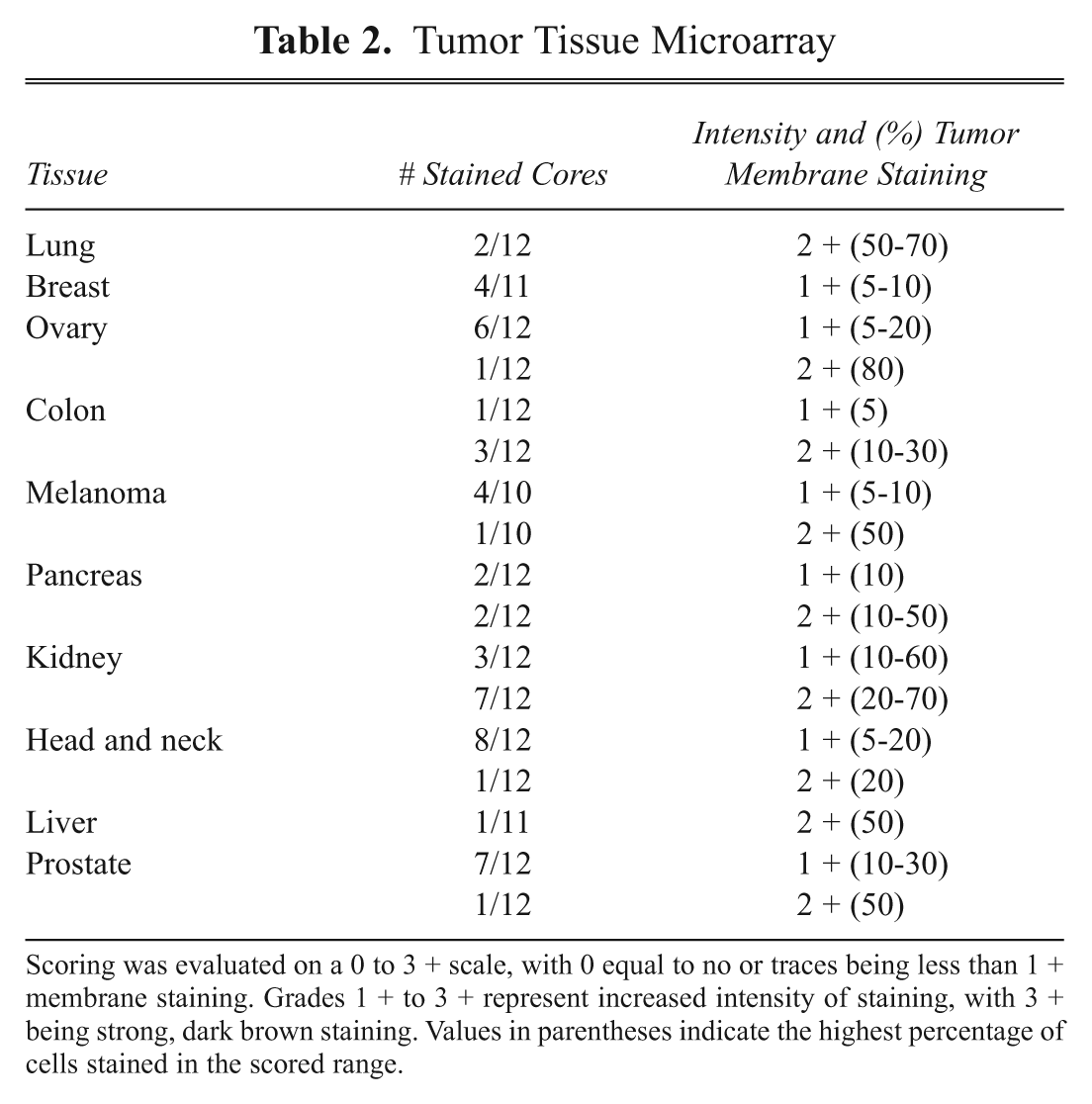
Scoring was evaluated on a 0 to 3 + scale, with 0 equal to no or traces being less than 1 + membrane staining. Grades 1 + to 3 + represent increased intensity of staining, with 3 + being strong, dark brown staining. Values in parentheses indicate the highest percentage of cells stained in the scored range.
Flow cytometry demonstrated that VB6-314-PE was reactive to most tumor cell lines tested. The strongest binding was to head and neck (SCC-25), pancreatic (BxPC-3), and breast (MDA-MB-435S) tumor cells, closely mirroring the pattern of reactivity observed with tumor tissue ( Table 3 ). The dissociation constant of VB6-314-PE was determined by flow cytometry using MDA-MB-435S cells and calculated to be 2.9 × 10−8 M (data not shown). However, this number may represent an underestimation as the same method measured a KD for VB6-845-PE that was almost 10 times lower than that obtained by BIAcore analysis. 15,16
VB6-314 Tumor Cells Profiling Data
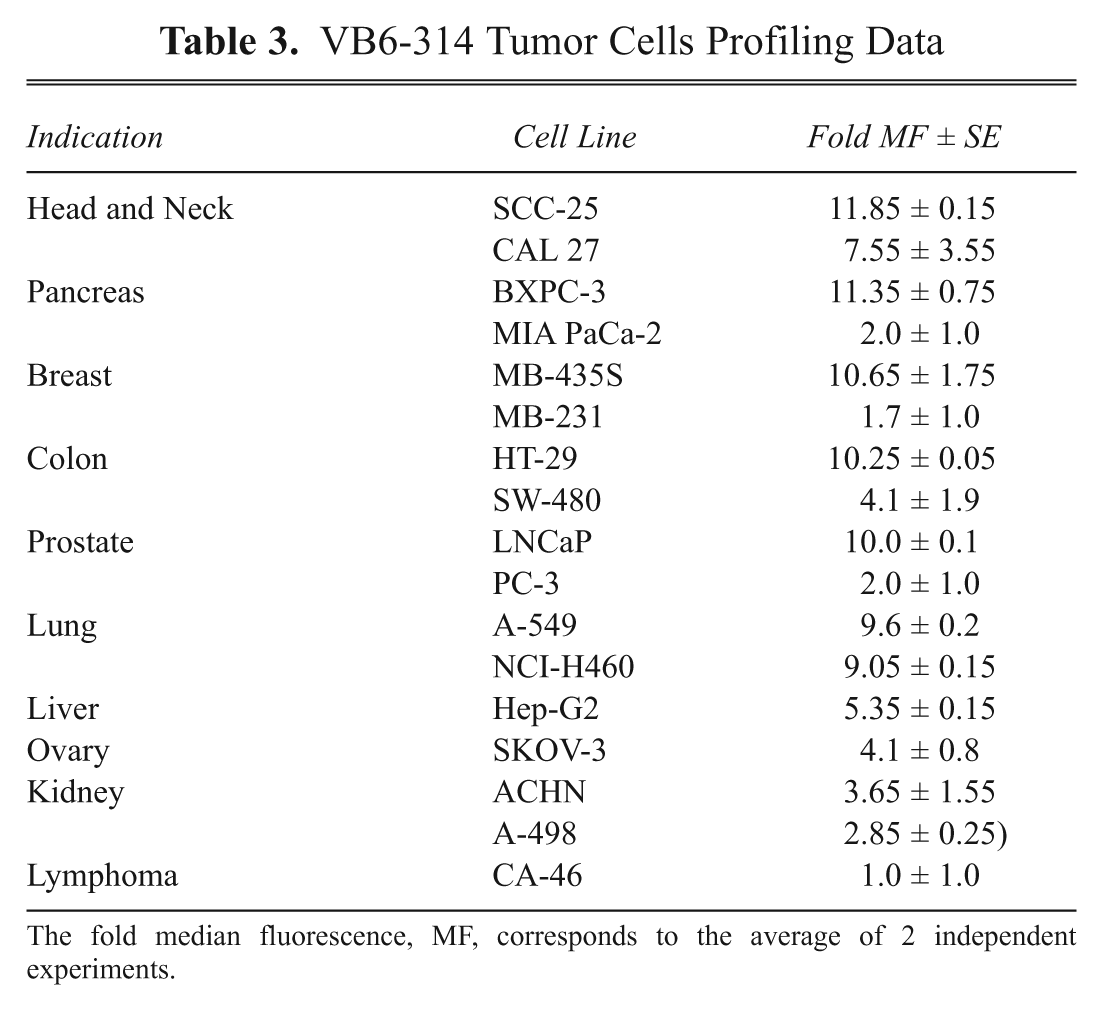
The fold median fluorescence, MF, corresponds to the average of 2 independent experiments.
Discussion
This report describes the development of the Fusogenics screening platform in which a Fab-PE immunotoxin format was used to select internalizing tumor-specific Ab fragments using a functional cytotoxic assay. The screening parameters were established with an internalizing anti-EpCAM Fab-PE fusion protein as a proof of concept. Subsequently, a Fab-PE immune library was created, and the specificity and selectivity of the process was illustrated with the biological characterization of clone VB6-314-PE.
The concept of the Fusogenics approach is based on the direct selection of internalizing Fab fragments. The Fab format was chosen over the scFv because of the inherent stability provided by the mutual interaction across the interface of the VL/VH and CL/CH domains and by the interchain disulphide bond. 17 Furthermore, many scFv fragments derived from natural antibodies are prone to dimerization and aggregation due to an exposed hydrophobic patch of amino acids normally buried in the interface of the variable and constant domains. 18 Dimerized or aggregated scFvs could trigger cell surface receptor internalization via cross-linking, leading to the selection of false positives. Therefore, the stability of the Fab ensures the selection of Ab fragments that do not require target dimerization for internalization. For the toxin moiety, the highly potent truncated form of PE was an ideal choice as only few molecules are required to kill a tumor cell. Since PE induces apoptosis by preventing protein synthesis, the readout of the Fusogenics approach was to measure an apoptotic index. 13 Although only a Fab-PE format has been tested, it is conceivable that any ligand, encompassing a stable antibody format or scaffold protein, could be engineered as a soluble fusion-PE protein. Similarly, the PE moiety could be changed to other types of effector proteins as long as they trigger a measurable biological effect.
Compared to other antibody discovery platforms, the Fusogenics approach does not permit a rapid de-convolution of the library for binder enrichment. To address this issue, 3 parameters were considered to minimize the size of the library without compromising its diversity. First, the library was created from enriched plasma B cells and thus focused only on antibody-producing B cells. Second, plasma B cells were obtained from regional draining lymph nodes of cancer patients with the intent of selecting for tumor antigen-driven B cells, thereby increasing the probability of identifying unique antibody fragments specific to cell surface TAAs. 19 Third, IgMs were omitted from the library by using IgG-specific primers during the PCR amplification. Combining these 3 characteristics created an enriched and diverse library of tumor-reactive clones.
One-third of the clones did not express a full-length Fab-PE fusion protein probably due to the mal-folding during heavy and light chain pairing, unfavorable codon usage, or stop codons introduced by the PCR reactions. Similar observations have been reported in particular with phage display where poorly folded scFv or Fab fragments yielded low numbers of phage, which are subsequently subtracted in favor of better producers during the panning selection. It is known that the coexpression with periplasmic chaperones could facilitate the folding of antibody fragments, leading to an increased level of soluble protein. 20 Therefore, it is conceivable that this approach could be applied to the Fusogenics concept to further increase the frequency of expressing clones.
The sequence analysis validated the immune library by demonstrating that the percentages of VH and VL subfamilies mirror the human B cell V-gene repertoire and was similar to that obtained from various phage libraries. 7 As well, the evenly distributed CDR3 heavy chain length and the lack of redundancy were indicative of a diversified immune library. During an immune response, somatic mutations are introduced into rearranged Ig genes, and as a consequence, B cells with improved antibody/antigen binding properties are selected. 21 The higher somatic mutation rates, found in the CDR1 and CDR2 loops compared to the framework, implied that antibody fragments of antigen-activated B cells were included in the immune library creation. A similar increase of somatic mutations in the CDR1 and CDR2 loops was reported with a phage library using isolated splenic B cells. 22
To date, all tumor-reactive clones selected with the Fusogenics approach are nonreactive with critical normal tissues showing the efficiency of the negative selection step, which eliminated the nonspecific binders created by the random VH-VL pairing. It may also reflect the fact that immune tolerance minimizes the appearance of self-reactive clones, thereby favoring those clones that are immunologically driven by the disease. Half of the clones that passed CN-TMA had reactivity restricted to 2 or 3 tumor indications, whereas the remaining antibodies, including VB6-314-PE, reacted with multiple tumor types and demonstrated higher levels of membrane reactivity. Indeed, the membrane binding and internalization of VB6-314-PE was confirmed by flow cytometry using a panel of epithelial tumor cells that express the antigen in its natural conformation. The double-digit nM dissociation constant obtained with VB6-314-PE is in the range of other selected clones (data not shown) and corresponds to the trend observed with phage immune libraries as the innate immune response generally yields antibodies with an average affinity. Xenograft studies have shown that scFvs with nM affinities are optimal for accumulation in a tumor, whereas higher affinities can actually impede diffusion into the tumor. 23 In addition, other factors such as the nature of the target, its density, and rate of internalization also affect the potency of an immunotoxin and hence in vivo efficacy. 24,25 Therefore, the need for an affinity maturation step will be determined on an individual basis, being guided by the preliminary data obtained from in vivo efficacy studies using tumor xenografts. In the event that affinity maturation is required, libraries containing randomized targeted CDR mutations will be engineered, and using the Fusogenics screening method, clones with higher cytotoxicity will be selected as a surrogate for improved affinity. This approach was validated with one of our preclinical candidates showing a 7-fold increased binding affinity (manuscript in preparation). Of note, the immunogenicity of the PE bacterial protein has limited the clinical benefit of PE-containing immunotoxins. 14 Therefore, for systemic applications, the final preclinical candidate will be engineered with the Fab fragment genetically linked to a variant protein of bouganin, a ribosome inactivating protein that has been de-immunized by T cell epitope depletion. 16
The Fusogenics approach complements other methods that select internalizing antibodies. Indeed, a similar strategy using PE38 has been described using purified full-length antibodies. 26 For this method, the ZZ domain derived from Streptococcal protein A is linked to PE38, and the ability of the Ab-ZZ-PE38 complex to kill tumor cells is determined. Approaches using a pH-sensitive fluorescent probe such as CypHer5, coupled to an antibody, also predicted internalization. 27 As well, a variation on phage display has been used to isolate phage from within the endosome as a means to select internalizing antibody fragments. 28 This strategy led to the identification of several internalizing scFvs that bind to TAAs such as MCAM/CD146. 29
In contrast to conventional discovery platforms, Fusogenics can potentially isolate antibodies against any target, known or novel, as long it is seen by the patient’s immune system. Not knowing the identity of the target could represent a challenge in drug development as this may raise concerns as to the appropriateness and design of toxicology studies and ultimately the safety of the molecule heading into clinical trials. To this end, we have developed a rapid proteomic approach for antigen identification based on immunoprecipitation with the antibody prior to 2D-LC separation in tandem with tandem mass spectrometry (MS/MS). 30
In conclusion, the Fusogenics approach permits the isolation of internalizing antibody fragments with proven cytotoxic potential. The selection stringencies are based on specificity to tumor tissues and selectivity against normal tissues, internalizing properties, and nM range affinity. And finally, since immunotoxins are generally produced by microbial expression, Fusogenics represents a streamlined development path from screening to preclinical evaluation with only a molecular reengineering step required to switch from the immunogenic PE toxin to the de-immunized bouganin payload.
Footnotes
Acknowledgements
We gratefully acknowledge Dr. Adrian Schwartz Mittelman and Shauna Loewen for technical assistance.
Jeannick Cizeau, Marianne Torres, Sharla G. Cowling, Stacy Stibbard, Arjune Premsukh, Joycelyn Entwistle, and Glen C. MacDonald are employees of Viventia Biotechnologies, Inc. who have direct financial interest in the subject matter discussed in this manuscript.