
Abstract
Bcl-2 family proteins are key regulators of apoptosis associated with human disease, including cancer. Bcl-2 protein has been found to be overexpressed in many cancer cells. Therefore, Bcl-2 protein is a potential diagnostic target for cancer detection. In the present study, the authors have identified several Bcl-2 binding peptides with high affinity (picomolar range) from a 5-round M13 phage display library screening. These peptides can be used to develop novel diagnostic probes or potent inhibitors with diverse polyvalencies.
Introduction
T
Overexpression of Bcl-2 prevents cells from undergoing apoptosis in response to a variety of stimuli. 3 This antiapoptotic property of Bcl-2 suggests that it may function as an oncogenic protein in developing tumors. In normal physiology, Bcl-2 is expressed in the ductal epithelia of the normal breast. 5-7 However, Bcl-2 proteins are overexpressed in many cancers, including 90% of colorectal adenocarcinomas; 30% to 60% of prostate, 70% of breast, and 20% of non–small cell lung cancers; and 60% of B cell–derived lymphomas. 8,9 Although the biological significance of Bcl-2 overexpression for tumor development and progression remains to be further evaluated, Bcl-2 protein-level changes are closely associated with histopathological features of tumors. 10 Therefore, Bcl-2 level may be a potential predictor of positive clinical outcome. In this sense, development of a specific diagnostic probe against Bcl-2 protein may result in a therapeutic agent for cancer early treatment.
High specificity and affinity of probes is the fundamental core of a successful diagnostic system. 11 Chemical/physical stability is required to apply these probes to a wide spectrum of diagnostic methods. 12 Although antibodies have been extensively used as a standard platform for biological detection system, 13 accumulating reports have identified nucleic acids or peptides as alternative probes with comparable (or higher) binding and specificity. Phage display of randomized peptide libraries is a standard technology for selecting peptides that target specific molecules, and this approach has been extensively used in various pharmaceutical biotechnologies. 14-17 Highly diverse libraries are constructed by fusing degenerate DNA fragments to one of the coat protein genes, which is encapsulated into the phage particles that display the encoded polypeptides on their surfaces. Library members with desired binding specificities and affinities can be isolated by in vitro biopanning. 18
In this study, we used an M13 phage peptide library and discovered diverse peptides that bind to Bcl-2 protein with a high affinity (picomolar range).
Materials and Methods
Materials
The Escherichia coli strain, BL21(DE3), was used as an expression host to produce Bcl-2 protein. Bacteria culture media, Bacto-tryptone and Bacto-yeast extract, were purchased from Difco (Becton Dickinson and Company, Franklin Lakes, NJ). An M13 peptide library screening kit (Ph.D. phage display peptide library kit) was obtained from New England Biolabs (Beverly, MA). Antibodies specific for monoclonal anti-M13, polyclonal glutathione S-transferase (GST), anti-Bcl-2, and anti-Bax were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All other chemicals used in this study, including isopropyl-1-thiogalactopyranoside (IPTG), were obtained from Sigma (St. Louis, MO).
Purification of GST-fusion protein
Construction and purification of recombinant Bcl-2 protein (rBcl-2) were performed as described elsewhere. 8,19 Briefly, the mature form of Bcl-2 protein was subcloned into pGEX-4T-2 plasmid (Amersham Bioscience, Piscataway, NJ) at EcoRI and XhoI sites (pGEX-4T2-Bcl2), so that the target protein was expressed as a GST-fused protein. The plasmid was transformed into BL21(DE3) E. coli bacteria and grown at 37°C in 1 L of Luria-Bertani (LB) medium containing 100 µg/mL ampicillin to an OD600 of 0.7. The cells were then induced to overexpress the target by the addition of 0.5 mM IPTG at 37°C for an additional 4 to 5 h. Cells were harvested by centrifugation, and the resulting pellet was frozen overnight at −80°C. Cells were resuspended in 10 mL of phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) and treated with lysozyme (0.5 mg/mL) at 4°C, with stirring for 30 to 60 min, followed by ultrasonication. After centrifugation at 15,000 g for 15 min at 4°C, insoluble particles were removed by filtration (0.45-µm syringe filter). The resulting supernatants were loaded onto a GST-Sepharose column (Amersham Bioscience), which was preequilibrated with PBST buffer (PBS containing 0.5% Triton X-100). The column was then washed with at least 3 bed volumes of PBST buffer, followed by PBS washing (5 bed volumes) to remove excess Triton X-100. Bcl-2 was eluted by a linear gradient of 20 mM glutathione (GSH, reduced form), 0.15 M NaCl, and 50 mM Tris (pH 8.0). Fractions containing target protein were collected and subjected to a desalting column to remove GSH.
Preparation of GST-free protein
GST-free Bcl-2 (rBcl-2) was generated by thrombin treatment according to the manufacturer’s instructions (Sigma). The cleavage reaction was terminated by the addition of 1 mM benzamidine. GST-free protein was purified by an ionic exchange column, Mono Q (Amersham Bioscience), with a 0.5 M NaCl linear gradient, and desalting column against 25 mM Na-phosphate, pH 7.4. Cleavage of GST-Bcl-2 was confirmed by Western blot using 2 different antibodies that specifically recognize the GST-tag (anti-GST) and Bcl-2 (anti-Bcl-2), respectively.
Peptide library screening
The Ph.D.-12 Phage Display Peptide Library (PhD12; New England Biolabs) with 2.7 × 109 peptide complexity was used for screening. Purified Bcl-2 protein, 0.5 µg, was immobilized onto a polystyrene plate (SPL) via hydrophobic interactions, and wells were then covered with 2% bovine serum albumin (BSA; 2% bovine serum albumin in Tris-buffered saline [TBST] containing 50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% Tween-20) for 1 h at room temperature. The wells were washed 3 times with TBST and used for biopanning. A specified amount of phage was added to the wells and incubated for 1 h at room temperature with gentle shaking. Wells were washed 10 times with TBST, and bound phages were eluted in a chemical elution (0.2 M glycine-HCl [pH 2.2], 1 mg/mL BSA). The eluates were then neutralized with 1 M Tris-HCl (pH 9.1). In subsequent rounds of biopanning (second to fifth rounds), the concentration of detergent or salt in TBST was increased as follows: 0.3% Tween-20 (second round), 0.5% Tween-20 (third round), and 0.5% Tween-20 plus 500 mM NaCl (fourth and fifth rounds, respectively). At the final round, bound phages were eluted by addition of excess amount of Bcl-2 protein. Eluted phages at each round were amplified using E. coli ER2738 strains to make sufficient copies for subsequent rounds of biopanning. After being propagated for 5 h, the bacterial cells were removed by centrifugation at 11,000 g for 15 min. The phages were partially purified by precipitation of the culture supernatant with polyethylene glycol (20% [w/v] polyethylene glycol-8000 and 2.5 M NaCl) according to the manufacturer’s protocol. After centrifugation at 10,000 rpm for 30 min, phage pellets were resuspended in TBS buffer (50 mM Tris-HCl [pH 7.5] and 150 mM NaCl) and used for the next round of biopanning.
DNA sequencing
An overnight culture of E. coli ER2738 was diluted 1:100 in LB medium and divided into 1-mL volumes, into which an individual phage displaying a Bcl-2 binding peptide was added. Cells were incubated at 37°C for 5 h, and then the supernatant containing the phages was harvested by centrifugation. Then, 200 µL of PEG/NaCl (20% [w/v] polyethylene glycol-8000 and 2.5 M NaCl) solution was added to precipitate the phages, and the resulting pellet was suspended in iodide buffer (containing 10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 4 M NaI) followed by alcohol precipitation. Single-stranded phage DNA was recovered and dissolved in dH2O. The single-stranded phage DNA sequences were determined with an automatic sequencing service (Macrogen, Inc., Seoul, Korea) using the -96 pIII sequencing primer. The amino acid sequence of the peptides was deduced from the genetic code information supplied by New England Biolabs.
Estimation of binding affinities on enzyme-linked immunosorbent assay
Phages displaying Bcl-2-binding peptides were prepared and their concentrations determined by measurement of plaque-forming unit (pfu) per volume. To estimate the binding affinities of peptides, the phages displaying Bcl-2 binding peptides were added to a 96-well plate containing Bcl-2 protein (0.5 µg/well) in a concentration-dependent manner, as indicated. The wells were then washed 10 times with TBST and probed with an anti-M13 antibody (mouse monoclonal antibody, 1:5000 in TBST, 0.2 µg/mL; Amersham Bioscience) for 1 h at room temperature. After washing with TBST buffer 5 times, antimouse IgG–horseradish peroxidase (HRP) conjugate was added and incubated for 1 h, followed by a color-developing reaction with TMB substrate solution (3, 3′, 5, 5′-tetramethylbenzidine [TMB]/ H2O2; Chemicon, Billerica, MA). After 15 min, the reaction was terminated by the addition of 1 M H2SO4, and the optical densities were measured with a Biotrak multiwell plate reader (Amersham Bioscience) at 450 nm. Alternatively, serial dilutions of biotinylated/or FITC-labeled peptides were incubated with Bcl-2 proteins immobilized on 96-well plate for 2 h. The biotinylated peptide bound to the target proteins was detected by incubation with streptavidin-HRP antibody (1:1000 dilution in TBST; BD Pharmingen, San Diego, CA). For FITC-labeled peptide, the binding affinity was determined by a fluorescence microplate reader (Molecular Devices, Sunnyvale, CA) using the soft Max V5 system, with excitation at 495 nm and emission at 520 nm.
Analysis of peptide binding to Bcl-2 in a mammalian cell system
A human cervical carcinoma cell line (HeLa) was transfected with a biotin-labeled peptide (Biotin-IHWSWTTVERPH) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). After incubation for 24 h, cells were lysed in buffer containing 40 mM Tris-HCl (pH 8.0), 120 mM NaCl, 0.1% NP-40, and proteinase inhibitor, and total cell extracts were incubated with streptavidin-conjugated agarose beads at 4°C for 30 min. The beads were recovered by centrifugation and were washed 3 times with the same cell lysis buffer. The proteins coupled to the beads were released by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer and were analyzed by Western blotting using a polyclonal antibody against Bcl-2. Coprecipitation of biotin-labeled peptides and Bax with streptavidin beads/HeLa cells (107 cells) lysed in 200 µL lysis buffer were incubated with biotin-labeled peptide for 60 min. Coprecipitation was performed with streptavidin beads. Western blot analysis of Bax (15% SDS-PAGE) was performed with the anti-Bax polyclonal antibody.
Peptide binding to cellular Bcl-2 protein was monitored by adding FITC-labeled peptide (10 ng) into HeLa cells under a fluorescence microscope. 20 HeLa cells were digested with 0.25% trypsin and grown on a 24-well chamber slide overnight. The Bcl-2 siRNA (sc-29214) was purchased from Santa Cruz Biotechnology. A control siRNA (sc-37007; negative control comprising a scrambled sequence with no significant homology to any known human gene sequences) was also purchased from Santa Cruz Biotechnology. One day before transfection with siRNA, HeLa Bcl-2 cells were plated on 12-well plates and grown in the appropriate medium supplemented with 10% serum and without antibiotics until a confluence of about 30% to 50% was reached. Cells were transfected with 100 pmol of Bcl-2 siRNA or 25 pmol of control siRNA per well in serum-free medium for 4 h and then cultured in the presence of serum (10%) and without antibiotics at 37°C for 48 h. Cells were washed 3 times with PBS and fixed with acetone at 4°C for 20 min before analysis. P6 peptide labeled with FITC was incubated with cells. PBS, different-sequence peptide control FITC, and siRNA were used as negative controls. After being washed 3 times with PBS, the slips were observed using a fluorescence microscope (Olympus, Melville, NY).
HeLa cells treated with 200 µM FITC-labeled peptide were washed twice with ice-cold PBS before fixation with ice-cold methanol. After blocking with 2% BSA in PBS containing 0.2% Triton X-100, cells were incubated with primary antibody against Bcl-2 for 1 h. Cells were washed with blocking solution 3 times and incubated with the secondary antibody conjugated with Alexa 594 (Molecular Probes, Eugene, OR) for 1 h. After washing 3 times with PBS, coverslips were mounted onto microscope slides using ProLong antifade mounting regent (Molecular Probes). The slides were analyzed using a fluorescence microscope.
Results
Expression and purification of Bcl-2 protein
To screen Bcl-2-binding peptides, we first prepared N-terminally GST-tagged wild-type Bcl-2 from bacteria. The Bcl-2 protein was expressed as soluble protein and purified to homogeneity by a single pass of GSH-Sepharose affinity chromatography. The purified Bcl-2 protein showed high homogeneity on 15% SDS-PAGE ( Fig. 1 ). Then, GST-fused Bcl-2 protein was challenged with thrombin digestion to remove N-terminal GST protein. The GST-Bcl-2 protein was originally expressed as a fusion protein containing a thrombin site between the target protein and the GST tag. After thrombin digestion, GST-free protein was further purified by ionic exchange column, Mono Q, in which 2 major peaks were observed and identified by Western blot using anti-GST and Bcl-2 antibodies. Finally, GST-free Bcl-2 was concentrated and confirmed by Western blot analysis ( Fig. 2 ).
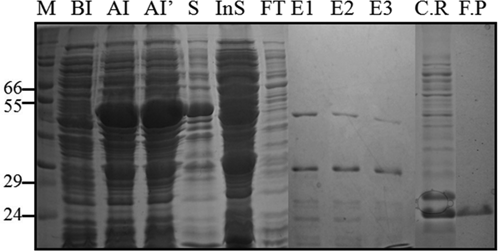
Preparation of Bcl-2 protein. Cells harboring pGEX-4T-2-Bcl-2 were induced to protein overexpression with 0.5 mM isopropyl-1-thiogalactopyranoside (IPTG) at 37°C for 4-5 h (BI, before induction; AI, after induction for 4 h; AI′, after induction for 5 h). Cells were resuspended in phosphate-buffered saline (PBS) buffer containing lysozyme and lysed by sonication. Supernatant was loaded onto a glutathione (GSH)–Sepharose column, and the protein was eluted by a linear gradient (elution buffer: 20 mM GSH, 150 mM NaCl, 50 mM Tris [pH 8.0]), and fractions (E1-E3) were resolved on 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Fractions containing rBcl-2 were collected and subjected to load on a desalting column to remove GSH (running buffer: PBS buffer). The GST-Bcl-2 was treated with thrombin to obtain GST-free Bcl-2 protein, and 2.5 mg of GST-Bcl-2 was challenged with 5 U of thrombin/mg of protein thrombin for 1 h at room temperature. The reaction was terminated by the addition of 1 mM benzamidine. The cleavage product was further purified on a Mono-Q anion-exchange column, which was preequilibrated with PBS, and eluted by 0.5 M NaCl linear gradient. The GST-free Bcl-2 protein was purified to high homogeneity in fraction F.P. M, molecular weight marker; S, supernatant after lysis; InS, insoluble protein (dissolved pellets in SDS-PAGE sample buffer); E1-E3, elution form bead; F.T, flowthrough; C.R, cleavage results from thrombin treatment; F.P, final product from Mono-Q column.
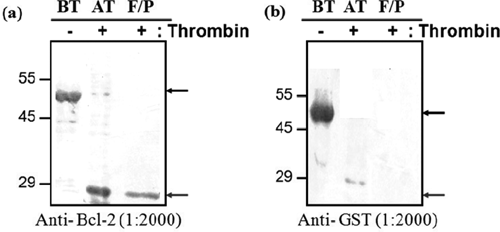
Western blot analysis of purified glutathione S-transferase (GST)–free Bcl-2 protein. Cleavage was evaluated by Western blot using (
Peptide library screening
We used an M13 phage display library to screen for peptides that specifically bind Bcl-2 protein. We isolated the M13 phages displaying Bcl-2-specific binding peptides as outlined in Figure 3 . We first immobilized Bcl-2 protein onto a polystyrene plate (SPL) and added the M13 phage library displaying random 12-residue peptides that were fused to the N-terminus of the gpIII protein. After 5 cycles of biopanning, we obtained 25 M13 phages and determined their DNA sequences. Based on amino acid sequences deduced by the DNA sequencing, the phages could be categorized into 7 groups according to their amino acid sequence similarities ( Table 1 ).
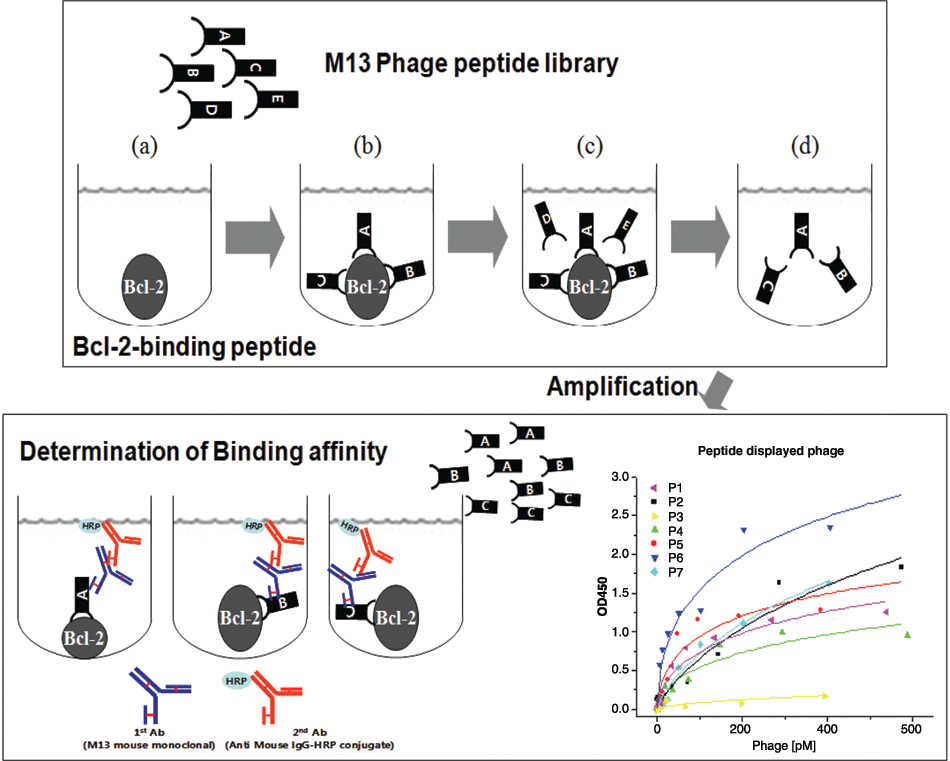
Schematic diagram representing the strategy used to screen M13 phage displays for Bcl-2-binding peptides. To identify Bcl-2-binding peptides with a high affinity, Bcl-2 protein was immobilized onto a plate, where the phage library was then added. After 5 rounds of biopanning, the phages displaying high-affinity Bcl-2-binding peptides were collected, and the peptide displayed on each phage was characterized.
Bcl-2-Binding Peptides Displayed on M13 Phages a
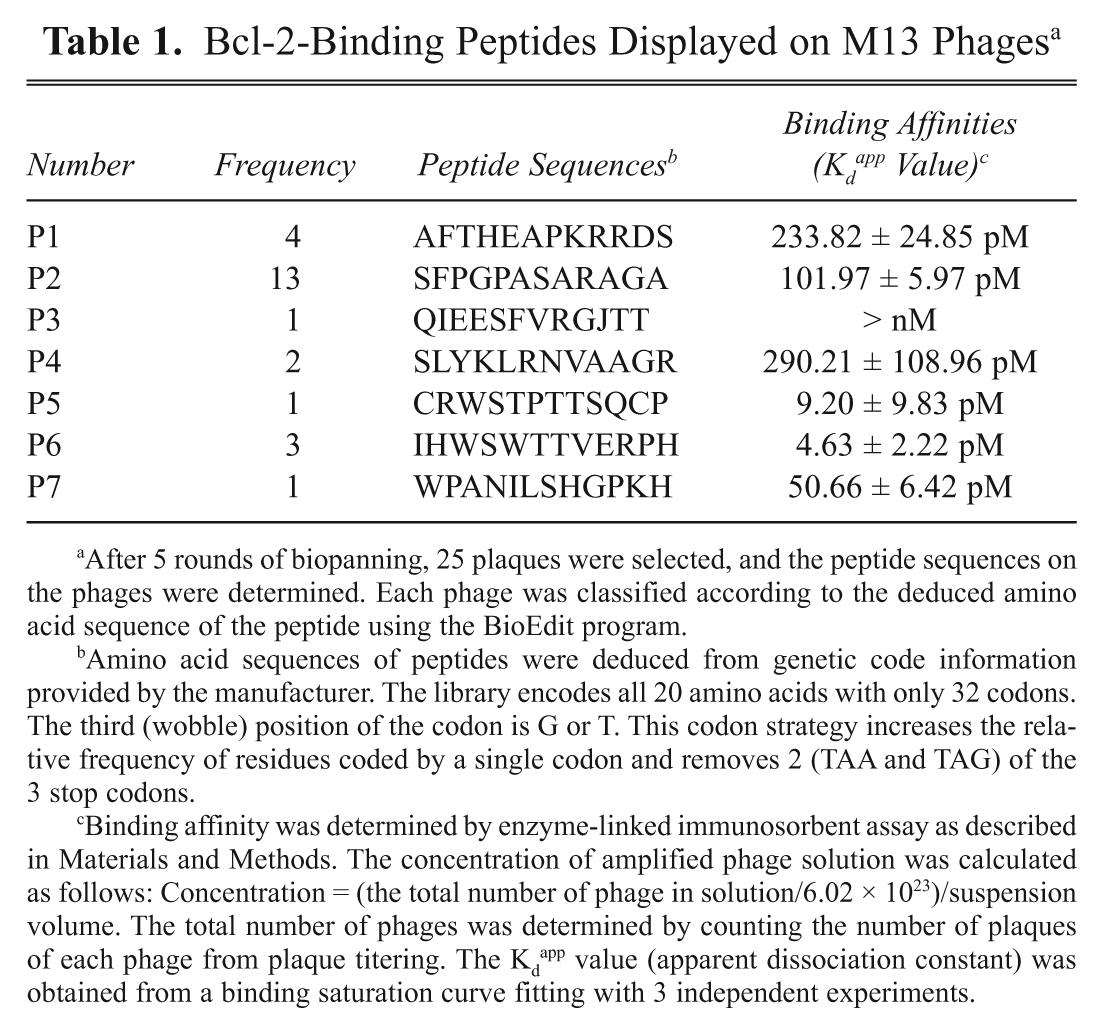
After 5 rounds of biopanning, 25 plaques were selected, and the peptide sequences on the phages were determined. Each phage was classified according to the deduced amino acid sequence of the peptide using the BioEdit program.
Amino acid sequences of peptides were deduced from genetic code information provided by the manufacturer. The library encodes all 20 amino acids with only 32 codons. The third (wobble) position of the codon is G or T. This codon strategy increases the relative frequency of residues coded by a single codon and removes 2 (TAA and TAG) of the 3 stop codons.
Binding affinity was determined by enzyme-linked immunosorbent assay as described in Materials and Methods. The concentration of amplified phage solution was calculated as follows: Concentration = (the total number of phage in solution/6.02 × 1023)/suspension volume. The total number of phages was determined by counting the number of plaques of each phage from plaque titering. The Kd app value (apparent dissociation constant) was obtained from a binding saturation curve fitting with 3 independent experiments.
The selected phages displaying Bcl-2-binding peptides were further characterized to estimate their binding affinities for Bcl-2 protein. The phages were first amplified and purified. The concentration of each phage was determined by titering serial dilutions of the purified phage solution. To determine binding affinities, Bcl-2 proteins were immobilized onto a plate, and then each phage was added in a concentration-dependent manner. After 30 min of incubation at room temperature, the wells were extensively washed, and the remaining phages were examined by enzyme-linked immunosorbent assay (ELISA) using the M13 antibody, which specifically recognizes the gp8 coat protein on the M13 bacteriophage. As shown in Table 1 , most of the phages showed comparable binding affinities (Kd app) for Bcl-2 protein in the picomolar (10−12) range except for P3 (nanomolar range). Among these, P6, which displayed the lowest Kd app value, was synthesized as either a biotinylated (Biotin) or FITC-labeled (FITC) peptide and was further characterized in vitro.
Characterization of P6 peptide (IHWSWTTVERPH) for Bcl-2 binding
To confirm P6 peptide binding to the Bcl-2, Biotin- and FITC-labeled P6 peptides were synthesized in vitro, and the binding affinities were examined by ELISA (
Table 2
). The binding affinity of this peptide was in the micromolar range (FITC-P6, 1.06 ± 0.43 µM; Biotin-P6, 9.62 ± 3.40 µM). Notably, the Kd
app of the peptide increased 2 to 3 orders of magnitude compared with the value on the phage-displayed P6 peptide. Concentrated peptide displays on the gP3 M13 tail coat protein phage (5 copies of this coat protein are structurally placed in close proximity) might be one of reasons for this difference. Next, FITC-labeled P6 peptides were tested for Bcl-2 binding in a cell system. HeLa cells, which are human cervical carcinoma cells, were treated with FITC-conjugated peptides, and P6 peptide addition produced high green fluorescence cell staining, whereas in control cells, HeLa cells incubated with FITC and different-sequence peptide were hardly shown in this staining (
Kd of the Biotinylated and FITC-Labeled Bcl-2-Binding Peptides

The dissociation constant (Kd) was obtained from a binding saturation curve fitting from 2 independent experiments.
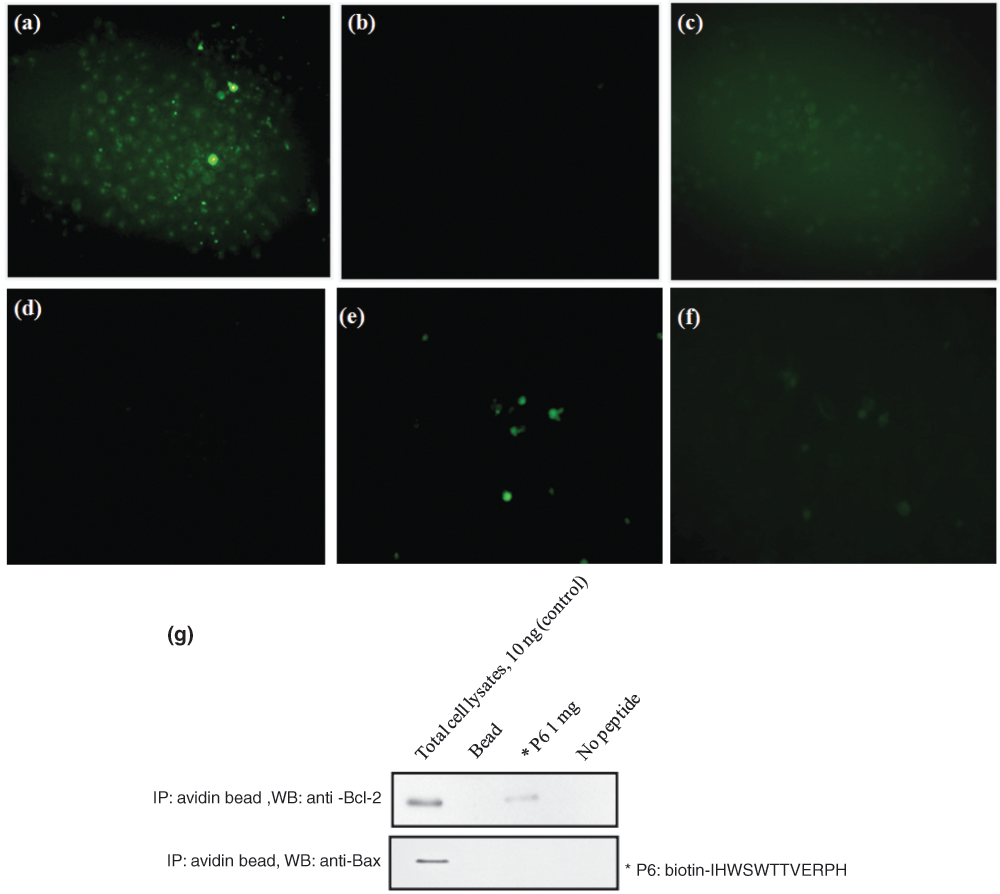
Incubation of FITC- or Biotin-conjugated Bcl-2-binding peptide (FITC-P6 and Biotin-P6) with human cervical carcinoma cells (HeLa). To determine if fluorophore-labeled P6 binds to Bcl-2 in cell system, HeLa cells were incubated in the (
In addition, P6 peptide binding to cellular Bcl-2 protein was determined by streptavidin pull-down, followed by Western blotting. Biotin-P6 peptide precipitated Bcl-2 from cell lysates when streptavidin beads were added to the samples ( Fig. 4g ). Binding of biotin-P6 peptide to Bcl-2 was examined by Bcl-2 Western blotting analysis of this pull-down assay. In contrast, Bax was not precipitated by biotin-labeled peptide. As shown in Figure 4g , cellular Bcl-2 protein was pulled down with the biotin-P6 peptide, confirming the specificity of this peptide probe toward Bcl-2 in a biological system. Moreover, analysis by fluorescence microscopy clearly revealed the colocalization of Bcl-2 by merging the FITC-P6 peptide ( Fig. 5a ) and Bcl-2-specific antibody ( Fig. 5b ; yellow pattern in Fig. 5c ). In addition, results from analysis of the specificity test, by fluorescence microscopy, revealed that the Bcl-2 siRNA treatment decreased the signal of these cells ( Fig. 4e ) since the cell labeling was decreased when compared to the control siRNA ( Fig. 4c ) treatment and FITC-P6 peptide ( Fig. 4a ).
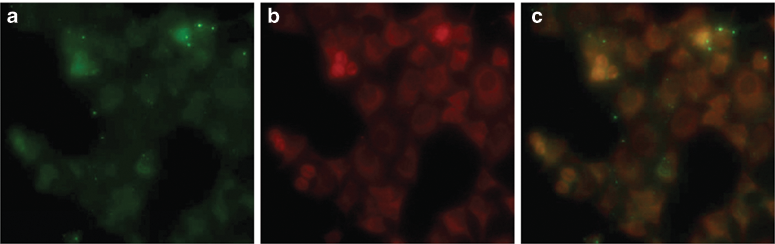
FITC-conjugated P6 peptide binds Bcl-2 in vivo. (
All together, our data indicate that this peptide can be a potential probe for in vitro diagnostics as well as in vivo imaging.
Discussion
Multicellular organisms eliminate redundant or damaged cells by cell death, termed apoptosis. 21 Apoptosis is controlled by a signaling equilibrium between prosurvival and proapoptotic pathways, such that disturbed apoptosis may contribute to the development of cancer and autoimmune and degenerative diseases. 22-26 Members of the Bcl-2 family act as master regulators of this cell death program. In particular, Bcl-2 protein activity is an antiapoptotic member of the Bcl-2 family. Aberrant Bcl-2 protein activity often renders cancer cells resistant to apoptotic signals, and the overexpression of Bcl-2 protein has been observed in many types of human cancer. 27 Therefore, approaches that alter the balance between prosurvival and prodeath Bcl-2 family members can have a potential benefit in the early stage of cancer treatment. 28 In addition, development of a specific diagnostic probe against Bcl-2 protein may contribute to successful cancer treatment.
In this study, we used phage display library screening to identify several Bcl-2 binding peptides. Peptides have several advantages over antibodies or other small molecular probes for therapeutic and diagnostic purposes. Because the peptides are 10 to 50 amino acids in length, they are chemically and physically more stable than antibodies and can more strongly and specifically interact with their targets compared to small molecular probes. We discovered 7 different Bcl-2 binding peptides (P1-P7), and most of them had comparable binding affinities in the picomolar ranges. Among these, P6 (IHWSTTVERPH) was further characterized in this study. Kd app of this peptide was in the micromolar range, which is 2 to 3 orders of magnitude higher than that of peptide displayed on the M13 phage. As mentioned in the Results section, several peptides were closely presented on the M13 phage particles. It can make the peptide on the phage more efficiently bind to Bcl-2. In this aspect, polyvalency can be one solution to improve the binding affinity of this synthesized peptide. That is, display of the peptide on a flexible polymer linker can mimic the molecular structure of the peptide on the M13 phage particle, thereby enhancing binding efficacy.
In addition, we showed that the P6 peptide specifically recognized binding to the Bcl-2 protein in the cell system (in vivo). FITC-labeled P6 peptide stained HeLa cells (
In conclusion, these results indicate that Bcl-2-binding peptide may be a useful diagnostic molecular reagent for detecting cancer.
Footnotes
Acknowledgements
This work was supported by the Korea Science and Engineering Foundation and Ministry of Education, Science and Technology through its National Nuclear Technology Program (2009-0081812).