
Abstract
The use of thermally denatured bovine serum albumin (tdBSA) as an additive in high-throughput screening (HTS) buffers has been studied with the aim of finding a surrogate to native albumin devoid of its inconveniences, in particular its compound masking effect. The presence of aggregates in the thermally denatured material did not have any negative impact on common readout technologies used in HTS such as fluorescence intensity (FLINT), fluorescence polarization, time-resolved fluorescence resonance energy transfer (TR-FRET) and luminescence. tdBSA rendered the same beneficial effects as native albumin in several assays or even improved its performance due to the lack of specific binding properties. Although tdBSA still binds compounds nonspecifically as any other protein does, it mitigates the compound masking effect observed with native albumin and can be postulated as a convenient surrogate to BSA for HTS purposes.
Introduction
A
Serum albumin is the most plentiful protein in blood plasma, where it contributes to osmolarity regulation. It is also widely recognized as a carrier protein by its unpaired ability to bind a wide diversity of molecules, from fatty acids to xenobiotics, due to the existence of 4 different binding sites in addition to 2 small metal binding pockets. 2 Such drug-binding properties make albumin a critical element influencing the in vivo effects of drugs, 3,4 and this is precisely the main caveat for using albumin as an additive in HTS: the effect of compounds in a given assay may be significantly masked, or even negated, upon binding to albumin. Although other proteins may be used as surrogates, they are also affected by important drawbacks such as poor solubility (casein) or a considerably higher cost.
It would, therefore, be desirable to have a protein available with the beneficial properties of BSA but devoid of its negative effects. Since BSA binding ability is dictated by its tertiary structure, thermally denatured BSA (tdBSA) may be a good candidate. However, solubility issues inherent to denatured proteins may be problematic as the tendency to aggregate may interfere with the optical detection technologies commonly used in HTS. In this article, we describe the best conditions to inactivate albumin by heating, check its performance in a variety of assay technologies, and evaluate its effect in real assays with particular attention to its possible effects on masking compounds.
Materials and Methods
Materials
Amplex™ UltraRed was from Invitrogen (Carlsbad, CA). 8-[[2-[(Fluoresceinylthioureido)amino]ethyl]thio]guanosine-3,′ 5′-cyclic monophosphate (8-Fluo-cGMP) was from Biolog (Bremen, Germany). Sheep anticyclic GMP polyclonal antibody was from Bethyl (Montgomery, TX). Bright-Glo™ reagents were from Promega (Madison, WI). Europium- labeled streptavidin was from PerkinElmer (Waltham, MA). Allophycocyanin-biotin (APC-biotin) was from Prozyme (San Leandro, CA). TAMRA-Crosstide and IMAP® beads were from Molecular Devices (Sunnyvale, CA). Enoyl-ACP reductase (FabI) from Plasmodium falciparum, isocitrate lyase (ICL) from Mycobacterium tuberculosis, human Akt kinase, human D–amino acid oxidase (DAAO), and human fatty acid synthase (FAS) were cloned, expressed, and purified from suitable vectors by the Biological Reagents department of GlaxoSmithKline following procedures published elsewhere. 5-9 All other reagents, including dansylsarcosine, fatty acid free albumin from bovine serum, β-lactoglobulin from bovine milk, and luciferase from Vibrio fischeri, were from Sigma-Aldrich (St. Louis, MO).
Preparation of thermally denatured BSA
A 100-µM solution of fatty acid free albumin from bovine serum in phosphate-buffered saline (PBS) was heated at 80°C for 15 min. The solution was then cooled down at room temperature for 2 h and subsequently stored at 4°C up to 1 month. For the studies on thermal denaturation, a 100-µM solution of BSA in PBS was heated at different temperatures for 30 min and then cooled down at room temperature for 2 h. Samples were centrifuged and the supernatant filtered (0.45 µm), and PBS was added to adjust the protein concentration as determined by absorbance at 280 nm.
Binding assay
Dansylsarcosine was used as a probe to monitor binding to BSA as already described. 10 The assay was carried out in 384-well polystyrene low-volume black plates (Greiner Bio-One, Gloucestershire, UK) in a final volume of 10 µL, including 2.5 µM BSA (concentration determined spectrophotometrically by the absorbance at 280 nm) and 5 µM dansylsarcosine. The binding of the probe was determined after a 10-min incubation period at room temperature, monitoring the increase in the fluorescent properties of the dansyl group upon binding to a hydrophobic environment: the emission at 530 nm was recorded after exciting at 360 nm in a Gemini SpectraMax™ plate reader (Molecular Devices). Then, 500 µM naproxen was used as a control of full dansylsarcosine displacement (Kd for naproxen was determined to be 1.6 µM in our lab).
Enzyme assays
The activity of FabI was determined by monitoring the consumption of NADH spectrophotometrically through the decrease in the absorbance at 340 nm at 25°C in 384-well clear-bottom polystyrene plates (Greiner Bio-One) using a 384-well plate spectrophotometer (SpectraMax Plus™; Molecular Devices). The assay media (final volume 80 µL) included 70 µM crotonoyl-CoA and 50 µM NADH in 50 mM sodium phosphate (pH 7.4).
The activity of ICL was followed by coupling isocitrate production from 300 µM glyoxylate and 500 µM succinate to isocitrate dehydrogenase (2 IU/mL) and diaphorase (0.2 IU/mL) in the presence of 250 µM resazurin and 1 µM NADH in 50 mM tricine (pH 7.4) with 5 mM MgCl2 and 0.4% (v/v) glycerol in a 20-µL assay volume. The increase of fluorescence intensity as a consequence of resorufin formation (λex 555 nm, λem 590 nm) was monitored continuously in 384-well polystyrene low-volume black plates (Greiner Bio-One) using a Gemini SpectraMax™ plate reader.
Akt kinase was monitored in the same black plates using 10 µM adenosine triphosphate (ATP) and 100 nM TAMRA-Crosstide in 50 mM HEPES (pH 7.5) with 1 mM dithiothreitol (DTT) and 10 mM MgCl2. The 10-µL reaction was stopped at different times by adding 10 µL of IMAP/® beads previously diluted at 1:500 from the commercial stock in assay buffer, and fluorescence polarization (FP) was measured (λex 535 nm, λem 580 nm) in an Analyst™ plate reader (Molecular Devices).
DAAO was measured in the same black plates using 2 nM enzyme and varying concentrations of several substrates (D-Ala, D-Pro, D-Ser, D-Met, D-Trp, and D-Phe) in 50 mM Tris buffer (pH 8.0) containing 250 nM flavin adenine dinucleotide. H2O2 production was monitored continuously with 1 IU/mL horseradish peroxidase and 100 µM Amplex™ UltraRed measuring resorufin formation as above.
Optical readouts
Dynamic light-scattering (DLS) measurements were performed on 60-µL samples in a 384-well clear plate (Cliniplate™; Thermo Fisher Scientific, Waltham, MA) using a NEPHELOstar Galaxy™ (BMG Labtech, Offenburg, Germany) plate reader equipped with a laser diode emitting at 635 nm.
The FP system used to check the effect of tdBSA consisted of a 10-µL mixture of 20 nM 8-Fluo-cGMP and a 1:200 dilution of commercial anti-cGMP antibody in 50 mM HEPES (pH 7.4) using the same black plates described above and monitoring FP (λex 485 nm, λem 535 nm). The time-resolved fluorescence resonance energy transfer (TR-FRET) system consisted of a 10-µL mixture of 10 nM APC-biotin and varying concentrations of Europium-labeled streptavidin in 50 mM HEPES (pH 7.5). TR-FRET was measured by exciting at 360 nm and monitoring emission at 620 and 665 nm. Luminescence was measured with the Bright-Glo™ system following the manufacturer’s instructions and using several concentrations of V. fischeri luciferase. In all these cases, readouts were performed on an Analyst™ reader.
Results and Discussion
Studies on BSA thermal denaturation
In earlier studies carried out on human and bovine serum albumins, aggregation was described as the main event responsible for denaturation only at high temperatures, whereas at mild temperatures, other processes were prevalent. 11 Since the presence of aggregates may be a nuisance for the optical detection systems typically used in HTS, it was decided to run a thermal stability study with BSA with the aim of finding the mildest conditions leading to full BSA inactivation as determined by the loss of ability to bind dansylsarcosine. Figure 1A shows a melting temperature (i.e., the temperature at which 50% of inactivation is obtained) of 64°C, with full inactivation being achieved at 80°C. Changes in the intrinsic fluorescence of BSA were also monitored to confirm that this loss of binding abilities was due to a significant conformational change and not to any other spurious effect. BSA contains 2 Trp residues that account for an emission peak with a maximum at 350 nm when excited at 295 nm, as shown in Figure 1B . This peak shifts upon heating to lower wavelengths with lower emission intensities, suggesting a change in the Trp environment to a more hydrophobic one 12 due to a gross conformational change that buries these residues or simply to aggregation following denaturation. To calculate the melting temperature from these spectral changes, the ratio of the emission intensities at 325 and 350 nm was used to normalize the total loss of intensity. From Figure 1C , a melting temperature of 67°C is deduced, in agreement with the functional data and therefore supporting the hypothesis that the loss of binding properties is due to conformational changes. As these data were obtained after having cooled the BSA solution for 2 h, the inactivation can be considered irreversible.
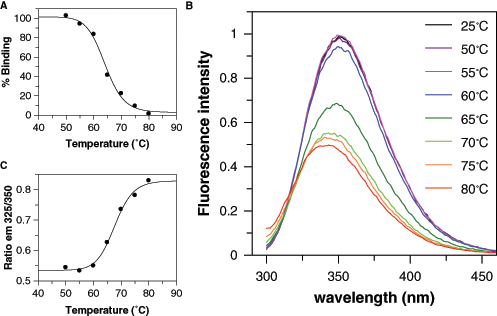
Thermal inactivation of bovine serum albumin (BSA). (
Kinetics of BSA thermoinactivation at 10 µM BSA followed first-order kinetics with a kinetic constant of 3.5 10–3 s−1 (data not shown). More concentrated BSA solutions yielded faster but more complex kinetic behaviors, with small light scattering observable in the preparation at wavelengths below 350 nm. Although no turbidity was visible to the naked eye and no appreciable sediment was obtained by centrifugation at 14 000 g, the presence of aggregates was evidenced by DLS measurements as explained below. The inclusion of N-ethylmaleimide (to prevent disulfide exchange with the unpaired Cys residue in BSA) did not change this behavior, whereas inclusion of urea to prevent aggregate formation resulted in a more thermostable protein that could not be fully inactivated after heating at 80°C for 30 min. Therefore, it was decided to inactivate BSA by heating a 100-µM solution in PBS at 80°C for 15 min (which, according to the kinetics described above, fully abolishes its binding properties). This material, referred to as tdBSA hereafter, was investigated for any interference with the optical detection systems typically used in HTS.
Effect of tdBSA on detection technologies
DLS experiments have evidenced that both BSA and tdBSA preparations include aggregates as deduced from the observed light scattering ( Fig. 2A ). Although these are more prevalent in the latter, their presence in the former is not negligible, and indeed the differences in light scattering when comparing BSA with tdBSA are similar to those when comparing BSA with plain buffer. Nonetheless, it was considered worthwhile to check the effect of these aggregates on readout technologies commonly used in HTS.
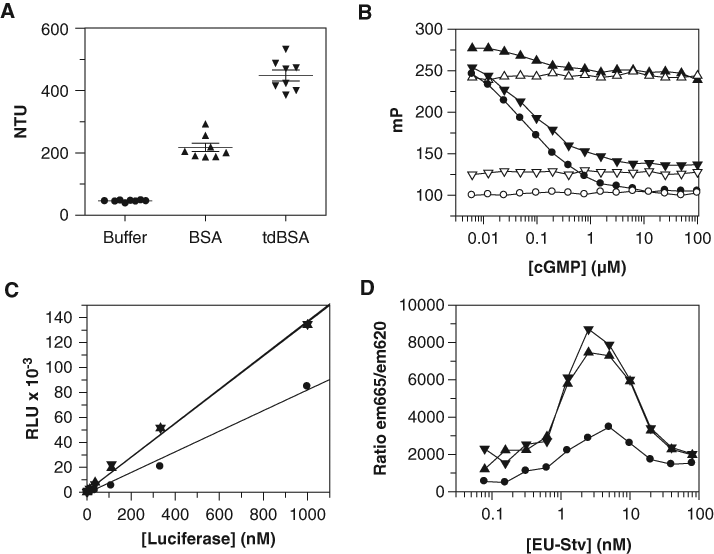
Effect of thermally denatured bovine serum albumin (tdBSA) on optical readouts. (
The fluorescent intensity (FLINT) of several fluorescent probes at different concentrations was checked in the presence and absence of 0.5% (w/v) tdBSA or BSA. Cy3B, 7-aminocoumarin, 7-hydroxy-4-trifluoromethylcoumarin (HFC), rhodamine 110, resorufin, 5-carboxytetramethylrhodamine succinimidyl ester (TAMRA), and fluorescein were tested. Both BSA and tdBSA caused some decrease in the slopes of the concentration-dependent FLINT curves (data not shown). The magnitude of such decrease ranged from 10% for Cy3B to more severe values for resorufin and HFC. Interestingly, the decrease was similar for both albumin preparations except for the most severe cases: BSA caused a 70% decrease in the fluorescence of resorufin and HFC, but the effect of tdBSA was more moderate (55% and 40% decrease, respectively). Since DLS values for tdBSA are higher than those for BSA, it is not possible to attribute the effect on fluorescence intensity to any optical interference caused by protein aggregates.
To test the effect on FP, the tandem formed by 8-Fluo-cGMP and an anti-cGMP antibody was used, and the ability of cGMP to displace the binding of the fluorescent probe to the antibody, hence decreasing the resulting FP signal, was evaluated in the presence and absence of 0.1% (w/v) BSA and tdBSA. Figure 2B shows that the use of BSA in this system is not permitted as the signal window disappeared due to the high background likely caused by the binding of 8-Fluo-cGMP to BSA, which was not prevented by cGMP up to 100 µM. However, tdBSA performed nicely in the assay: although the signal window was slightly reduced, the sensitivity window for cGMP remained unaffected (from 5 nM to 1 µM). Although in the case concerning this detection system, an HTS campaign could be run without any protein additive, it may serve to illustrate cases (particularly assays based on FP technology) where the inclusion of protein additives is necessary but the use of BSA is banned due to its ability to bind small molecules (e.g., a fluorescent probe). In such cases, tdBSA seems to be an excellent alternative.
The effect on luminescence was evaluated by running a titration of luciferase with the commercial Bright-Glo™ system: both BSA and tdBSA improved the performance of the system, as observed in Figure 2C . Noticeably neither BSA nor tdBSA caused any increase in the background (i.e., the signal detected in the absence of luciferase). Both preparations of BSA also caused a beneficial effect in the performance of the TR-FRET system shown in Figure 2D : a significant increase in the ratio of the intensity of emissions at 665 and 620 nm was noticeable, caused by an increase in the emission intensity at 665 nm, whereas that at 620 nm remained unaffected (data not shown). Although at this point it is not possible to justify the reasons for such improved behaviors, it seems likely that both preparations of albumin may stabilize some of the components of the assay or simply increase their effective concentration by preventing adsorption to plasticware. The bell-shaped curves observed are a consequence of the excess of Eu over APC when the former is above 10 nM, which increased the intensity of the emission at 620 without affecting that at 665 nm.
Effect of tdBSA on the performance of HTS assays
After having proven that tdBSA is not detrimental to the optical detection systems typically used in HTS, we investigated its performance as a BSA surrogate in assays already known to require albumin. Four assays encompassing different detection technologies were tested, all of them involving soluble enzymes as the screened target: FabI from P. falciparum (signal decrease assay monitoring absorbance), human Akt (signal increase assay monitoring FP), and human DAAO and ICL from M. tuberculosis (signal increase assays monitoring FLINT). Figure 3 shows how both albumin preparations affect the activity of the enzymes. The dependency of the initial rate on the concentration of FabI, ICL, and Akt is shown in Figures 3A , B , C , respectively: both albumin forms increased the specific activity of the 3 enzymes as deduced from the increase in the slopes of the plots. In the 3 cases, tdBSA behaved similarly to BSA, suggesting that both can be used indistinctly. A closer look at the changes in the slopes demonstrates that both albumins increased 1.5-fold the observed activity of FabI, circa 2-fold that of ICL and more than 6-fold that of Akt. Regarding DAAO, the study was extended to several substrates that are transformed by the enzyme with different catalytic efficiencies. The enzyme kinetic parameters were determined for these substrates in the presence and absence of BSA and tdBSA, and the values of the maximum velocities, which are dependent on the effective enzyme concentration, were compared. As shown in panel Figure 3D , there was an increase in the Vmax values when BSA or tdBSA was present, and this effect was independent of the substrate used, as noticed by the linear correlations obtained. From the slopes of these correlation plots (1.3 for BSA and 1.4 for tdBSA), it is deduced that both albumin preparations caused a significant increase in the effective concentration of DAAO in the assay medium. To summarize, in the 4 cases considered, the benefits procured by tdBSA were identical to or better than those procured by BSA and represent an increase of at least 40% in the “usable” enzyme (or even 600% in the case of Akt), therefore providing precious savings for HTS.
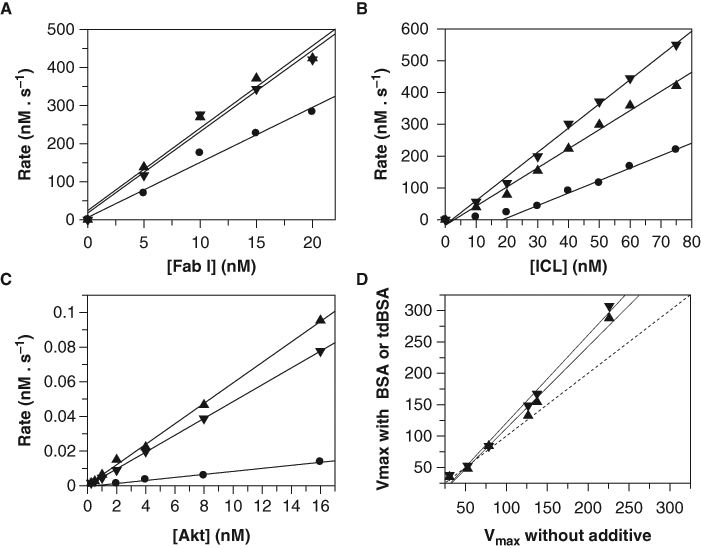
Effect of thermally denatured bovine serum albumin (tdBSA) on several assays. Titration of (
As detailed in the introduction, one of the major concerns about using albumin in HTS is based on its potential to mask the effect of compounds given its ability to bind them. We have therefore investigated the extent of this problem and whether the use of tdBSA may contribute to alleviating it. We have compared the potencies in the presence and absence of BSA and tdBSA of several hits found in different assays involving a diversity of targets: glucose 6-phosphate dehydrogenase, 5 cytochrome P450 isoforms (1A2, 2D6, 2C9, 2C19, and 3A4), 2 monoamine oxidase isoforms (A and B), and human FAS. Interestingly, from all these inhibitors, only those acting on FAS (which were closely related analogues belonging to the same chemotype) showed sensitivity to the inclusion of additive proteins in the assay buffer. Consequently, it may well be that the deemed compound masking effect is not as widespread as it is commonly believed, although it is obviously dependent on the chemical features of each individual compound or compound class. Figure 4 shows a significant drop in potencies for most of the circa 80 FAS inhibitors tested when either BSA or tdBSA was included ( Figures 4A and 4B , respectively), although the effect was more pronounced for BSA, probably because tdBSA had lost the ability to specifically bind some compounds. If this reasoning holds true, the drop in potencies seen in panel B must be due to nonspecific binding, and therefore the same phenomenon would be observed if any other protein was used instead. Indeed, Figure 4C shows that the values obtained in the presence of a totally unrelated protein such as β-lactoglobulin correlated acceptably with those obtained in the presence of tdBSA. Therefore, tdBSA mitigates the masking effect of BSA by abolishing the specific binding of compounds, although, as any other protein, it cannot prevent the negative effect caused by the unspecific binding intrinsic to its proteic nature.
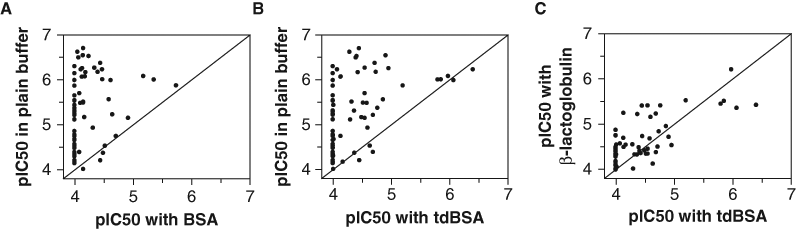
Effect of protein additives on the potencies of fatty acid synthase (FAS) inhibitors. Seventy-eight FAS hits, identified in a previous high-throughput screening, were tested against the enzyme as already described
13
and their pIC50s determined in plain buffer and compared with the values obtained in buffer with (
Footnotes
Acknowledgements
The authors thank María Jesús Vázquez for her help in executing and interpreting some of the experiments described in the manuscript.