
Abstract
Latent infection with Epstein-Barr virus (EBV) is a carcinogenic cofactor in several lymphoid and epithelial cell malignancies. At present, there are no small-molecule inhibitors that specifically target EBV latent infection or latency-associated oncoproteins. EBNA1 is an EBV-encoded sequence-specific DNA binding protein that is consistently expressed in EBV-associated tumors and required for stable maintenance of the viral genome in proliferating cells. EBNA1 is also thought to provide cell survival function in latently infected cells. In this work, the authors describe the development of a biochemical high-throughput screening (HTS) method using a homogeneous fluorescence polarization (FP) assay monitoring EBNA1 binding to its cognate DNA binding site. An FP-based counterscreen was developed using another EBV-encoded DNA binding protein, Zta, and its cognate DNA binding site. The authors demonstrate that EBNA1 binding to a fluorescent-labeled DNA probe provides a robust assay with a Z factor consistently greater than 0.6. A pilot screen of a small-molecule library of ~14,000 compounds identified 3 structurally related molecules that selectively inhibit EBNA1 but not Zta. All 3 compounds had activity in a cell-based assay specific for the disruption of EBNA1 transcription repression function. One of the compounds was effective in reducing EBV genome copy number in Raji Burkitt lymphoma cells. These experiments provide a proof of concept that small-molecule inhibitors of EBNA1 can be identified by biochemical HTS of compound libraries. Further screening in conjunction with medicinal chemistry optimization may provide a selective inhibitor of EBNA1 and EBV latent infection.
Introduction
E
At least 9 viral proteins and multiple noncoding RNAs have been detected in cells latently infected with EBV. Many of these have oncogenic potential when expressed ectopically in various model systems, 12 and some are essential for EBV immortalization of primary B cells in tissue culture. However, many of these viral oncogenes are not consistently expressed in EBV-associated tumor tissue where the viral genome can persist in a more quiescent state relative to the initial transforming process. In most EBV-associated tumor tissue, the viral genome is maintained through the consistent expression of the Epstein-Barr virus encoded nuclear antigen 1 (EBNA1). 13 EBNA1 is a sequence-specific DNA binding protein that binds to the EBV origin of plasmid replication (OriP) and facilitates the DNA replication and nuclear persistence of the viral genome in proliferating cells. 14-16 EBNA1 is consistently expressed in most, if not all, EBV-associated tumors and is required for the efficient establishment of EBV infection during B cell immortalization. Inhibition of EBNA1 by siRNA depletion 17 or by ectopic expression of a dominant negative mutant leads to loss of B cell survival. 18 EBNA1 binds to several sites in the EBV genome, where it is known to affect both viral chromosome stability and gene expression programs. 19-22 Recently, EBNA1 was also found to bind to several sites in the cellular genome, suggesting that it might alter host cells functions by directly interacting with chromosomal locations. 23
Small-molecule inhibitors of virus infection have been identified for numerous viruses, including herpesviruses. 24,25 Among these, acyclovir and phosphonoacetic acid derivatives have proven to be the most effective family of inhibitors of herpesvirus DNA polymerases and lytic cycle DNA replication. 25 Despite these potent inhibitors of DNA polymerase, there remain few therapeutic agents that are equally effective against latent infection of herpesviruses. EBV is a particularly attractive herpesvirus to target for latent infection because it expresses a large number of well-characterized proteins during latency and can be readily cultured as a latent virus. Because most EBV pathogenesis is associated with latent infection, identification of inhibitors of latent infection is of great clinical significance. Inhibitors of lytic infection are only marginally effective in the treatment of EBV-associated malignancy. A few chemotherapeutic anticancer agents have been found to inhibit EBV infections. Hydroxyurea has been shown to cause the loss of EBV genomes from some Burkitt lymphoma tissue and has been used effectively in the treatment of EBV-associated thymomas. 26,27 Because hydroxyurea is thought to act on the cellular enzyme ribonucleotide reductase, its inhibition of EBV is indirect and therefore subject to potential cellular toxicities and complications for long-term use. 28,29
A broadly used method for development of small-molecule inhibitors is to use high-throughput screening (HTS) of compound libraries to identify candidate lead compounds that can be further derivatized to improve medicinal and pharmacological properties. In this work, we describe the development of a robust fluorescence polarization (FP) HTS assay for small-molecule inhibitors of EBNA1 DNA binding. FP assays are ideal for HTS because they are homogeneous real-time assays that can be used to rapidly assess the binding properties in solution. 30 Moreover, FP assays have been used successfully to develop HTS for inhibitors of numerous enzyme-substrate interactions, as well as for identification of inhibitors of DNA binding proteins, such as C/EBP and Myc. 31,32 EBNA1 is an attractive target for small-molecule inhibitors because of its critical and consistent role in EBV-associated tumors. EBNA1 is also an attractive target because it is a viral-encoded protein with no known host-cell orthologue. Identification of small-molecule inhibitors of the EBNA1 DNA binding function will be a first step in the development of a selective inhibitor that is effective in cell-based and animal models of EBV tumorigenesis.
Materials and Methods
Expression and purification of recombinant EBNA1 DBD
Amino acids 459 to 607 of EBNA1, encoding the DNA binding domain (DBD), were expressed as a hexa-histidine fusion protein in Escherichia coli. Expression was induced in Rosetta2 cells with 0.3 mM isopropyl β-D-thiogalactoside (IPTG) for 3 h at 25°C. Soluble protein was recovered using a modified version of the method described by Frangioni and Neel 33 and purified via Ni-NTA agarose. Bound protein was extensively washed with 20 mM HEPES (pH 7.9), 1 M NaCl, 5 mM 2-mercaptoethanol, 40 mM imidazole, and 10% glycerol to dissociate nonspecific DNA bound to EBNA1 prior to elution in buffer containing 250 mM imidazole. Peak fractions of the eluted proteins were pooled and dialyzed against 20 mM HEPES (pH 7.9), 500 mM NaCl, 5 mM 2-mercaptoethanol, 10% glycerol, and 0.2 mM phenylmethylsulfonyl fluoride (PMSF).
Fluorescence polarization assay
A reaction mix was prepared containing 200 mM NaCl, 20 mM Tris-Cl (pH 7.4), 1 mM dithiothreitol (DTT), 10 µg/mL bovine serum albumin (BSA), and 10 nM Cy5-labeled EBNA1 BS hairpin with or without 246 nM purified recombinant EBNA1 DBD protein. This solution was incubated for 20 min at room temperature to promote the establishment of equilibrium binding of EBNA1 to the DNA hairpin prior to dispensing (BioTek MicroFlow Select; BioTek, Winooski, VT) 30 µL of this solution to each well of a 384-well microplate containing 0.5 µL of compounds dissolved in DMSO. Fluorescence polarization (EX: 620, EM: 680) was measured using an Envision Xcite Multilabel Reader (PerkinElmer, Waltham, MA).
Small-molecule screen
Chemical compounds that pass all Lipinski and drug reactivity filters used to qualify drug-like libraries and confirmed to be at least 80% pure by liquid chromatography/mass spectrometry (LC/MS) were obtained from the Lankenau Chemical Genomics Center (Wynnewood, PA). Compounds were preplated (0.5 µL) in 100% DMSO in columns 3 to 22 of 384-well black opaque Optiplates (PerkinElmer). The 14,000 compounds were screened as a mixture of 10 compounds per well. Each compound in the library was screened at a final concentration of 15 µM and is compressed in 2 independent dimensional arrays orthogonal to each other, such that each compound is represented twice surrounded by 9 different molecules in each assay well. DMSO (0.5 µL) was preplated in columns 1, 2, 23, and 24 of each assay plate. These wells were used for Maximum (DNA probe + protein) and Minimum (probe alone) controls of fluorescence polarization of EBNA1 DNA binding. To compound containing assay plates, 30 µL of a preformed EBNA1:Cy5-DNA hairpin complex was dispensed to wells using a BioTek MicroFlow Select. After a 1-h incubation at room temperature, FP (EX: 620, EM: 680) was measured using an Envision Xcite Multilabel Reader (PerkinElmer). Percent inhibition of EBNA1 DNA binding was calculated for each compound well relative to assay plate control wells—that is, % inhibition = (mPMax − mP Cmpd)/(mPMax − mPmin) × 100. Upon deconvolution, results were stratified into 4 categories: actives (i.e., the bioactive compound displays >75% inhibition of EBNA1 DNA binding and cleanly maps to a unique well in both the horizontal and vertical dimensions), ambiguous (i.e., the bioactive compound maps to 2 or more wells in either dimension), orphan (i.e., an orthogonal match cannot be identified in the second dimension), and inactive (<74% inhibition of EBNA1 DNA binding activity). This threshold cutoff was determined by averaging by normalized percent inhibition of all data points, plus 3 standard deviations of that average. This threshold cutoff yielded a hit rate of ~0.3% that was selected for confirmation. Chemicals selected for further analysis were reordered as powders from ChemDiv (www.chemdiv.com) and were designated as LB2 (#3241-0296), LB3 (#3241-0772), LB7 (#1071-0020), and SC7 (#K048-1003).
EMSA
An electrophoretic mobility shift assay (EMSA) reaction buffer was prepared containing 10% glycerol, 200 mM NaCl, 20mM Tris-Cl (pH 7.4), 1 mM DTT, 10 µg/mL BSA, 10 nM Cy5-labeled EBNA1 BS hairpin, and with or without 246 nM purified EBNA1 DBD. This solution was incubated for 20 min at room temperature to promote equilibrium binding. Then, 30 µL of this solution was dispensed to Eppendorf tubes containing 0.5 µL of a test compound in DMSO and mixed. Samples were then loaded onto a 6% polyacrylamide gel and electrophoresed for 90 min at 170V in 1× TBE. Nucleic acid migration was visualized using a Typhoon Imager (General Electric, Fairfield, CT).
IC50 calculations
To determine the relative IC50s of candidate hit compounds, an 11-point, 2-fold titration of each compound in 100% (v/v) DMSO was assessed in duplicate with an 80-µM compound concentration as the upper limit. Percent inhibition of EBNA1 DNA binding at each concentration of a compound was calculated relative to assay plate control wells—that is, % inhibition = (mPMax − mP Cmpd)/(mPMax − mPmin) × 100. Data from duplicate measurements were fitted separately to a 4-parameter fit logistic model to determine the relative IC50 concentration for each compound. GraphPad Prism 5.0 software was used to generate a 4-parameter fit dose-response curve.
Luciferase reporter assays for EBNA1 activity
293T cells (human embryonic kidney cells transformed with SV40 T antigen) were added to a 24-well plate at a concentration of 50,000 cells/well in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS). A Qp-Luciferase reporter construct was designed to assay the DNA binding function of EBNA1 in cell-based assays. The Qp region of EBV (nucleotides 49,712-50,250) were amplified by PCR and cloned into pGL3-Basic (Invitrogen, Carlsbad, CA) using Asp718-HinDIII restriction sites. Following an 18-h incubation at 37°C to allow cells to adhere, cells were transfected using 2 µL Lipofectamine (Invitrogen) per well. All the cells were transfected with 10 ng/well of a Renilla expression plasmid (N1457), 200 ng/well of a QP-Luciferase Reporter Plasmid (N1852), and 6.25 ng/well of a FLAG-EBNA1 (N803) or control FLAG (N799) vector. After 6 h, the transfection medium was replaced with fresh medium supplemented with test compounds added to achieve final concentrations ranging from 100 to 1.56 µM. Cells were incubated at 37°C for 48 h and then analyzed for luciferase activity using the Promega Dual Reporter system (Promega, Madison, WI).
EBV genome maintenance assay
Raji cells (EBV-positive Burkitt lymphoma–derived cell line obtained from ATCC, Manassas, VA) were grown at a density of 2 to 4 × 106 cells/mL in 2 mL of RPMI media supplemented with 10% FBS, 10 mM streptomycin, and 10 mM penicillin in 6-well plates. Test compounds dissolved in 100% DMSO were added to cultures to achieve a final concentration of 10 µM. Cells were grown at 37°C for 3 days and then passaged 1:10 into fresh media with the same concentration of drug for an additional 72 h. Genomic DNA was isolated using a ChIP lysis buffer/phenol chloroform method. DNA was quantified using quantitative PCR with primers for cellular actin and the DS region of the EBV genome.
Results
Protein preparation and purification
To develop a high-throughput biochemical assay for EBNA1 function, we expressed and purified the EBNA1 DBD as a hexa-histidine amino-terminal fusion protein. Hexa-histidine-tagged EBNA1 DBD (aa 459-607) was expressed in E. coli and purified over Ni-NTA agarose to near homogeneity (
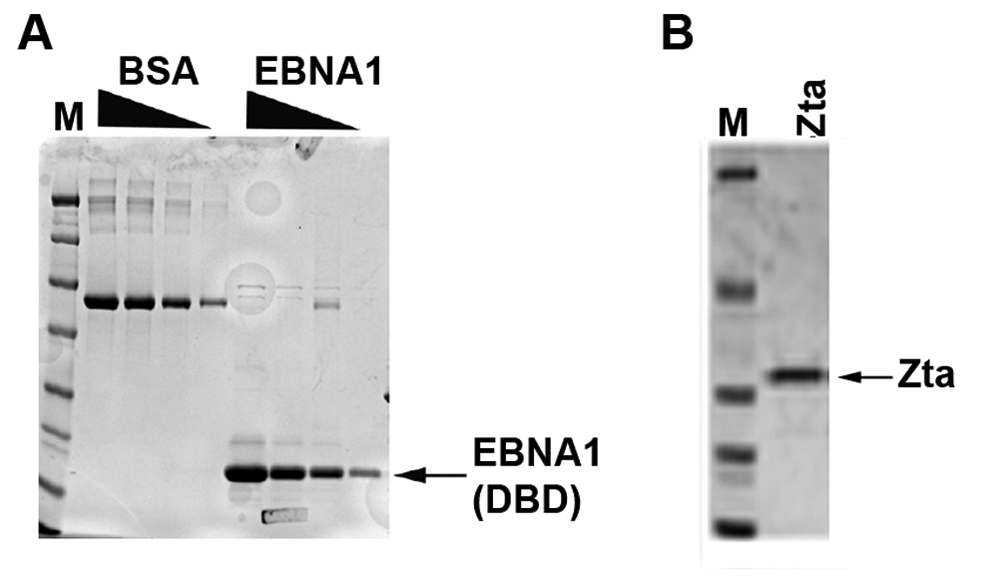
Protein purification of EBNA1 and Zta. (
Development of an FP assay for monitoring EBNA1 DNA binding
FP assays monitor changes in the molecular rotational properties of a polarized light in solution. As such, changes in the rotation rate of fluorescent molecules can be induced by altering the mass of the fluorescently labeled molecule, which can be readily detected by FP. We therefore attempted to develop an assay using a relatively small fluorescently labeled DNA probe bound by EBNA1. A similar assay was developed for Zta to use as a counterscreen to assess compound selectivity. For EBNA1 binding, a DNA probe was synthesized with a fluorescent Cy5 modification at the 5′ terminus. Because the consensus sites for EBNA1 are palindromic, we initially designed a self-annealing oligonucleotide containing an 18-bp perfect palindrome with the high-affinity consensus sequence from the OriP family of repeats (GGGTAGCATATGCTACCC).
34,35
EBNA1 bound this probe with high efficiency in EMSA (
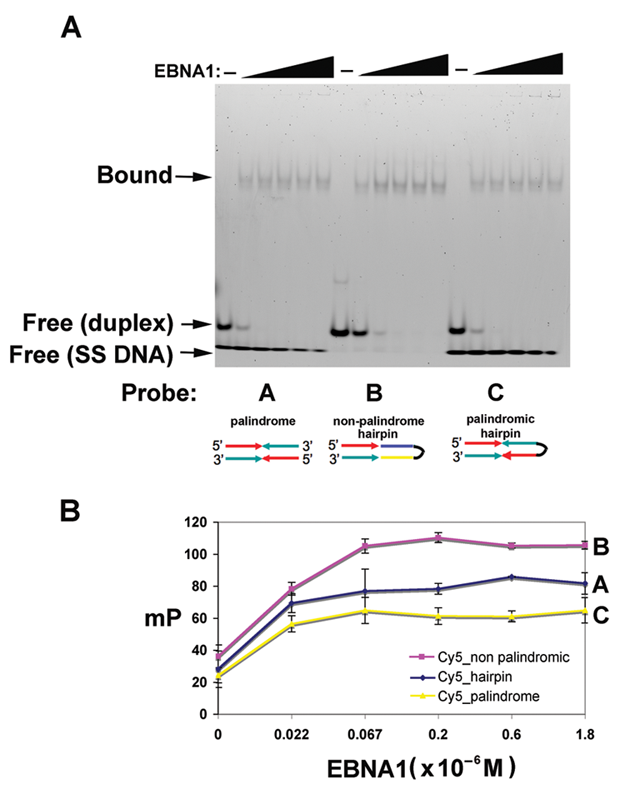
Development of an electrophoretic mobility shift assay (EMSA) and fluorescence polarization (FP) assay for high-throughput analysis of EBNA1-DNA binding. (
Assay adaptation to high-throughput format
The FP assay was miniaturized to a 384-well microplate format. During this process, we optimized the readout relative to a number of variables that included assay volume, fluorescent tracer concentration, EBNA1 concentration, DMSO sensitivity, assay signal stability, and reagent stability as a function of temperature and freeze-thaw cycles. In culmination, we assessed the optimized assay’s variation by calculating the Z factor for 2 plates performed independently on 3 successive days, where 192 wells from each plate contained either the Cy5-DNA hairpin probe alone or the EBNA1:Cy5-DNA hairpin complex in 1.5% DMSO.
36
In general, the replicate plate experiment for the EBNA1 FP assay yielded an average Z factor of 0.6, with all 6 plates scoring >0.55
HTS of a compound library
A small-molecule library of 14,000 highly diverse compounds with pharmacological properties in compliance with Lipinski’s rule of five
37
was selected for an initial screen to identify inhibitors of EBNA1 DNA binding in vitro. To increase cost- and time-effectiveness of the screening process, the library was plated in an orthogonally compressed format. The compression consisted of combining 10 compounds per well with each compound appearing in 2 different wells surrounded by 9 different compounds. This approach has proven to be highly efficient for large-scale screening campaigns.
38
From this screen, we identified ~40 compounds that displayed >75% inhibition at 10 µM concentration of compound (
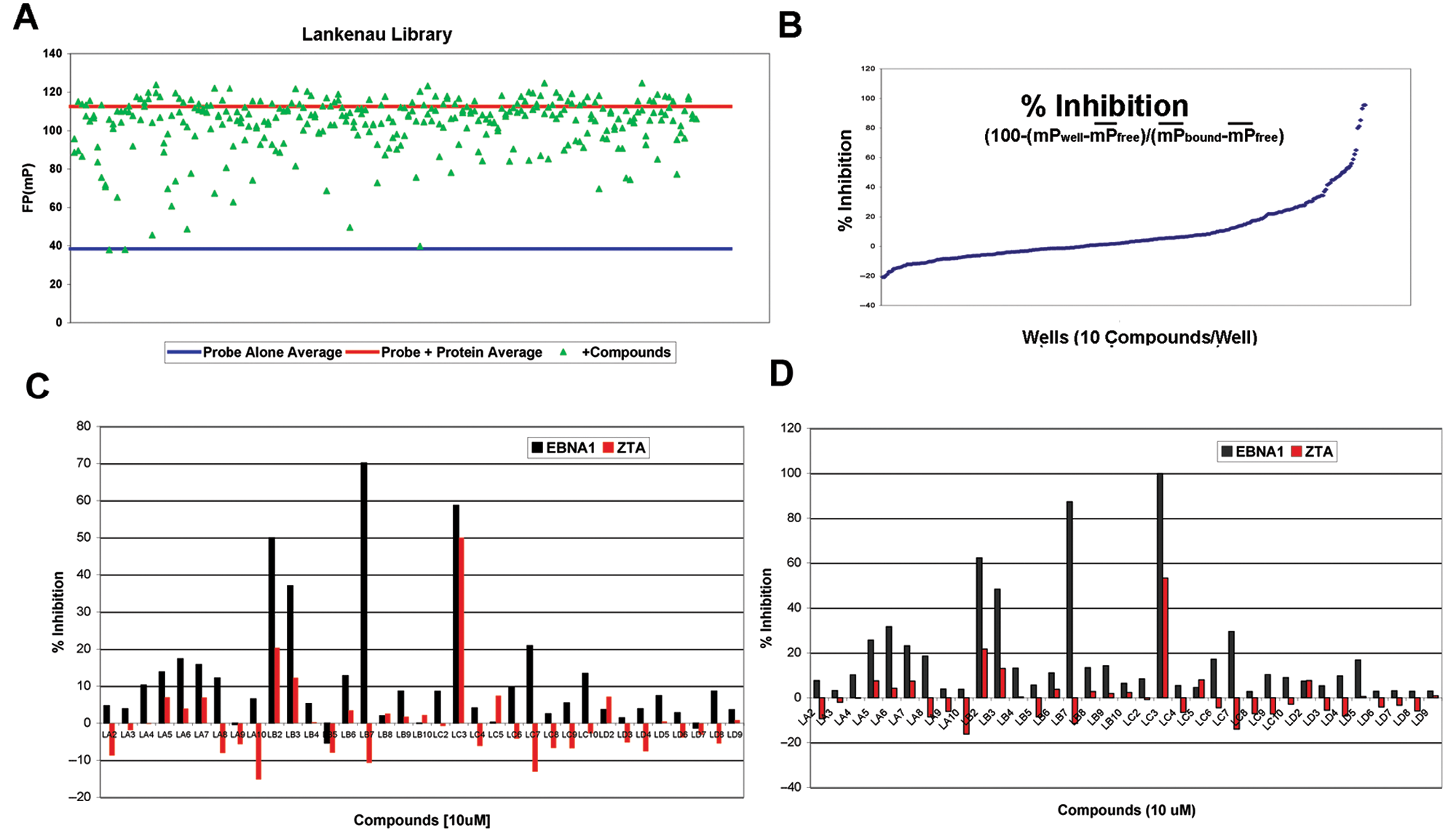
Summary of high-throughput screen of 14,000 compounds for inhibition of EBNA1-DNA binding. (
IC50 determination of candidate inhibitors
The chemical structures of the 4 compounds selected from the HTS and counterscreen are shown in
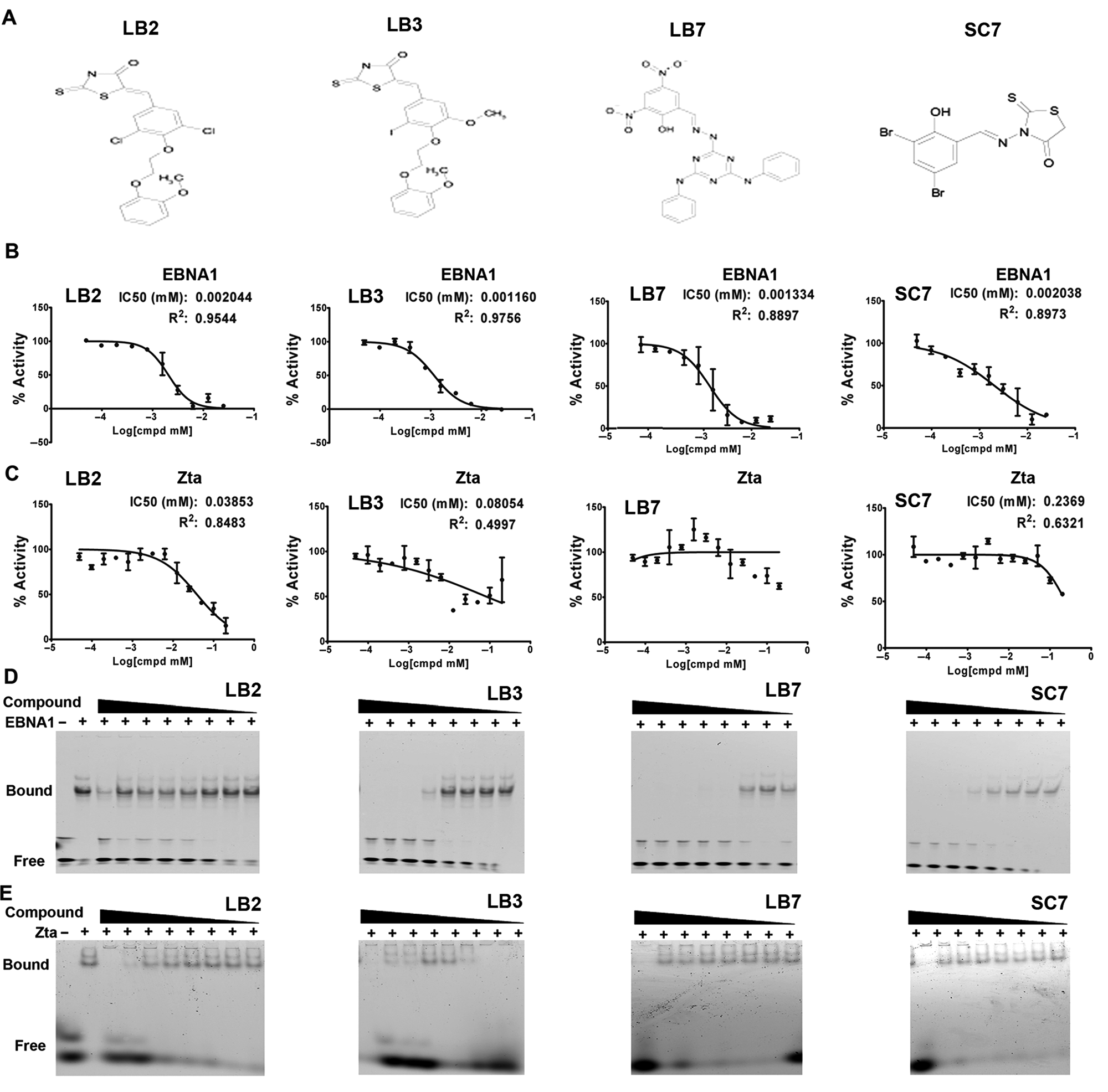
IC50 analysis of candidate inhibitors of EBNA1. (
Inhibition of EBNA1 transcription repression function in cell-based assays
LB2, LB3, LB7, and SC7 were tested in a cell-based assay for their ability to disrupt EBNA1 binding and function in live cells. EBNA1 is a potent transcriptional repressor of the Q promoter (Qp) because it binds directly to 2 sites positioned over the transcription initiation region (
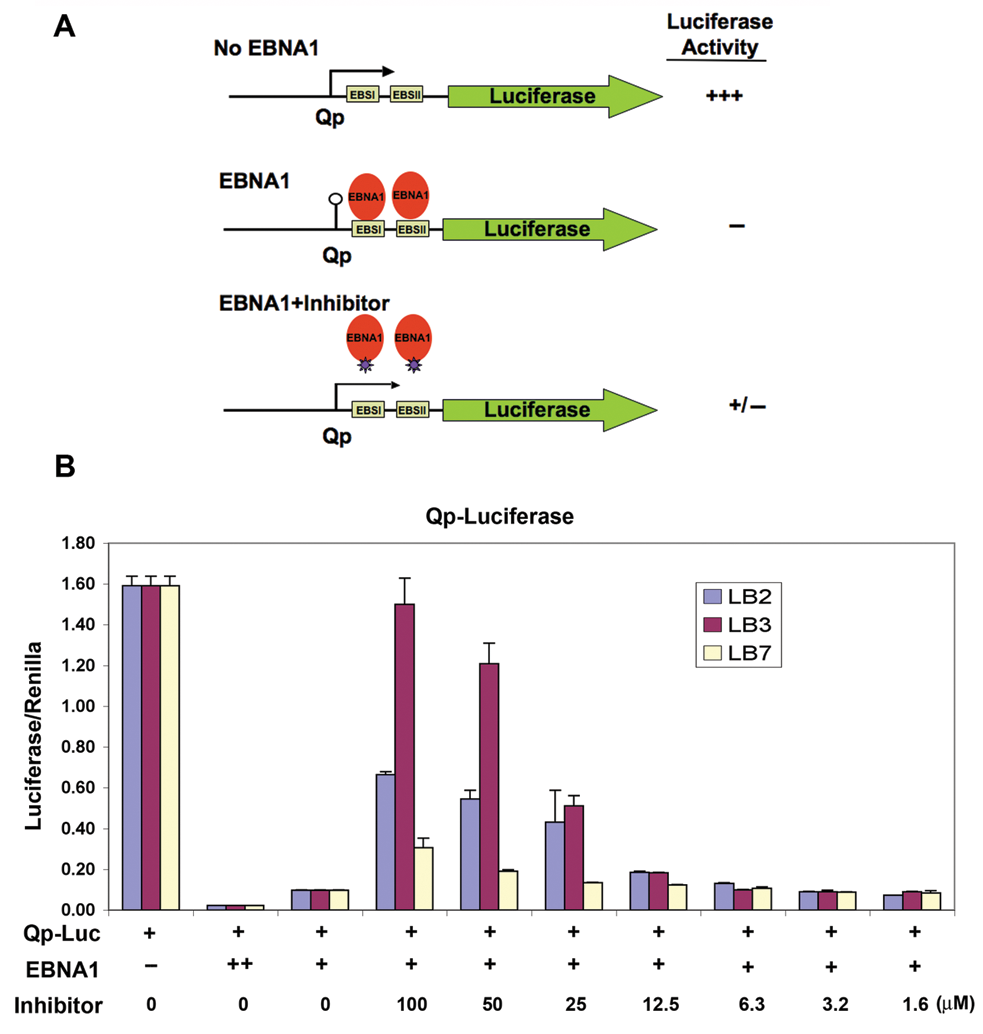
Inhibition of EBNA1 transcription repression in 293T cells. (
Elimination of latent EBV episomes
Candidate compounds were tested for their ability to deplete EBV genome copy number from latently infected Burkitt lymphoma cells. The Raji Burkitt lymphoma cell line carries ~100 copies of EBV per cell, but the copy number can be reduced by treatment with a known EBV inhibitor, such as hydroxyurea (HU).
29
We compared the ability of the 3 compounds LB2, LB3, and LB7, which had some anti-EBNA1 activity in cell-based assays, to function similar to HU in causing the elimination of EBV episomes (
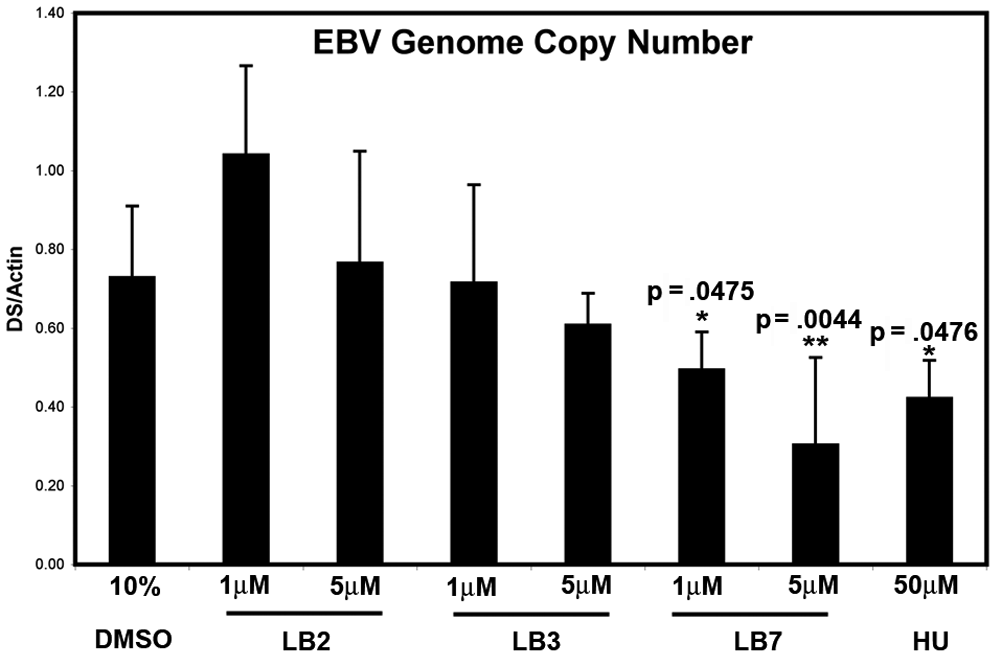
Reduction of Epstein-Barr virus (EBV) genome copy number in Raji Burkitt lymphoma cells. Raji cells were cultured with control DMSO; with 1 or 5 µM LB2, LB3, LB7; or with 50 µM hydroxyurea (HU) for 6 days (as indicated) and then assayed for DNA content by real-time PCR with primers specific to EBV DS region or cellular actin. EBV genome copy number is presented as the abundance of EBV (DS) relative to cellular (actin) DNA. Error bars were calculated as standard deviation from the mean. Significance relative to DMSO control was calculated using an unpaired t-test (*<.05, **<.01).
Discussion
In this work, we have developed methods to identify small-molecule inhibitors of EBNA1-DNA binding using biochemical assays. We screened a small compound library of ~14,000 molecules and identified ~4 candidates that showed selective inhibition of EBNA1 relative to a control DNA binding protein, Zta. Three of the compounds showed some activity in cell-based assays, and 1 compound was comparable to HU in the elimination of EBV genomes during long-term (6-day) treatment of EBV-positive Burkitt lymphoma cells in tissue culture. Although these findings suggest that methods are available for discovery of potential inhibitors of EBNA1, the candidate compounds identified are unlikely to be clinically relevant without significant modifications that enhance target selectivity and reduce cellular toxicity. Future campaigns with more extensive compound libraries would benefit from a combination of both biochemical and cell-based screening approaches. Furthermore, validation of the mechanism of inhibition using X-ray crystallography of the EBNA1-inhibitor complex will also provide critical information for enhanced inhibitor activity. Nevertheless, we demonstrate a proof of concept that small-molecule inhibitors can be identified for EBNA1 DNA binding, which could be of great value in the treatment of EBV-associated disease, as well as a research tool to control the functional binding of a high-affinity sequence-specific DNA binding protein. We anticipate that additional screening, combined with structure-activity relationship and medicinal chemistry, may provide an effective small-molecule inhibitor of EBNA1 for cellular and animal-based assays.
Footnotes
Acknowledgements
We thank Andreas Wiedmer and other members of the Lieberman lab for technical instruction and support. We acknowledge the Wistar Institute Cancer Center Core Facility for Protein Expression, Libraries, and Molecular Screening and the Core Facilities for Genomics and Flow Cytometry.
This work was funded in part by grants from NIH (3R21NS063906) to PML.