
Abstract
The search for new receptor ligands is important in the study of embryonic stem (ES) cell differentiation processes. In this study, a novel peptide (HGEVPRFHAVHL) with a specific ability to bind with neural stem cells derived from rhesus monkey ES cells was successfully screened out using a Ph.D-12 peptide phage display library. High affinity and specificity of the HGE phage were shown in an enzyme-linked immunosorbent assay. The binding ability of the phage could be matched with that of a chemically synthesized peptide with a sequence identical to that displayed by the phage, indicating that this binding capability manifests a peptide sequence. Combined with quantum dots, the HGE peptide can be used as a direct tool to show optical imaging of specific binding on a single cell membrane. Further results of Western blot showed that the HGE peptide interacted with 48/34-kDa proteins on the membrane of neural stem cells. This work is the first time that a phage display technique has been applied in ES cell differentiation studies. The findings extend the utilization of a targeting agent for neural stem cells and can also be used as a research tool in studying other cell lineages derived from ES cells.
Keywords
Introduction
E
However, little is known about the exact pathways involved in early human development or organogenesis, nor has the process been replicated in vitro using an ES cell model, indicating limitations in possible clinical applications. In the process of ES cell differentiation, because cells communicate with each other, as well as with their immediate niche through surface receptors, certain changes in the activities of these receptors are likely to be specified for particular lineages. Thus, for a certain cell lineage derived from ES cells, by identifying the responsible receptor ligands, we could label, isolate, and characterize the specific cell lineages and, furthermore, investigate relative pathways. Toward this end, an established phage display technology 5,6 has been used to selected peptide ligands with high affinity and specificity to neural stem (NS) cells derived from ES cells. The NS cells are uniquely suitable for the study of early human neural development and cell-based therapy. 7 Various peptides and antibodies have been discovered by panning whole cells both in vitro and in vivo by using the phage display strategy, 8-10 but the application of the phage display to identify special peptide ligands in ES cell differentiation has never been reported before.
Although most recent studies focus on human ES cells, clinical applications must be tested for appropriate animal models. 11 Therefore, choosing the primate’s ES cells that share the most common characteristics with human ES cells is a good option to avoid any ethical problems.
In this study, on the basis of a mature process of ES cell differentiation into NS cells, we applied a negative/positive biopanning procedure in suspending whole cells to enrich phage pools. Combined with quantum dots labeling, we show that the selected peptide is able to bind R-NS cells with high affinity and specificity.
Materials and Methods
Cell culture and preparation
Culture of R-ES cells
The rhesus monkey ES cell line RS366.4 (R-ES cells), provided by the Wisconsin Regional Primate Research Center (University of Wisconsin, Madison), was cultured on a feeder layer of pMEFs isolated from embryos of pregnant ICR mice, with an ES medium consisting of 85% Dulbecco’s modified Eagle’s medium (DMEM; Hyclone, Logan, UT), 15% fetal bovine serum (FBS; Gibco BRL, Carlsbad, CA), 1% nonessential amino acid (Gibco BRL), 1 mM L-glutamine (Gibco BRL), and 0.1 mM β-mercaptoethanol (Amresco, Solon, OH) at 37°C in the presence of 5% CO2.
Prior to the panning procedure, ES cells were quickly detached by dispase (5 mg/mL, Gibco BRL), washed twice with phosphate-buffered saline (PBS), and suspended in a blocking buffer (3% FBS in PBS) at 1 × 106 cells/mL.
RA-induced differentiation and isolation of neural stem cells derived from R-ES cells
Dispase (0.5%) digested R-ES cell clumps (50-100 cells) were placed in 0.6% agar-coated Petri dishes (Falcon, Becton Dickinson Labware, Franklin Lakes, NJ), grown in a culture medium the same as R-ES cells. After 6 days of embryoid body (EB) formation, cells were treated with 10−6 mol/L retinoic acid (RA). At 10 days, EBs were transferred into 1% gelatin-coated cell culture dishes (Falcon, Becton Dickinson Labware) with an NS cell–defined medium (DMEM: F12,50 × B27). After another 6 days of culturing in the defined medium, the neural tube-like rosettes with neural stem cells (R-NS cells) were isolated by incubating with 0.1 mg/mL dispase at 37°C for 15 min. The rosette clumps retracted, whereas the surrounding flat cells remained adherent. Then, clumps were collected and plated into a culture flask for 30 min to separate contaminated individual cells. Isolated cells were identified by immunocytochemistry.
Obtained R-NS cells were washed twice with PBS and suspended in a blocking buffer at 1 × 108 cells/mL. For biopanning, periphery cells that remained adherent in dishes after enzymatic treatment were collected by 0.25% trypsin–1 mM EDTA digesting, washed twice with PBS, and suspended in a blocking buffer at 1 × 106 cells/mL.
Immunocytochemistry and RT-PCR to identify neural stem cells
R-NS cells were plated in a 1% gelatin-coated 24-well plate (Falcon, Becton Dickinson Labware) with a B27-defined medium. After adherence, they were washed twice with PBS, fixed with 4% paraformaldehyde for 15 min at room temperature (RT), washed 3 times with PBS (5 min each), and incubated in a blocking buffer (1% bovine serum albumin [BSA] in PBS) for 2 h at 37°C. They were incubated with primary antibodies at 4°C overnight, washed 3 times with PBS, and incubated with secondary antibodies 1 h at 37°C. Primary antibodies and secondary antibodies were diluted with 1% BSA-PBS as described by the human neural stem cell characterization kit (CHEMICON, Temecula, CA). After final wash, cells were mounted in PBS and detected by Eclipse TE2000 fluorescence microscopy (Nikon, Tokyo, Japan). Images were processed with Image-Pro Plus software (Media Cybernetics, Bethesda, MD). Statistical analyses were performed using Image-Pro Plus software 6.3 (Media Cybernetics).
Total RNA was extracted by Trizol reagent (Invitrogen, Guangzhou, China). First-strand cDNA was synthesized according to the handbook of the RT-PCR kit (Takara, Dalian, China), and 1 µL of the cDNA reaction mix was subjected to PCR amplification with DNA primers selected for PCR amplification with DNA primers for genes OCT4 (forward, 5′-GACAACAATGAGAACCTTCA-3′; reverse, 5′-CACATCCTTCTCTAGCCCAA-3′), SOX2 (forward, 5′-CCCTGTGGTTACCTCTTCC-3′; reverse, 5′-CTCCCATTTCCCTCGTTT-3′), nestin (forward, 5′-AGAGGGGAATTCCTGGAG-3′; reverse, 5′-CTGAGGACCAGGACTCTCTA-3′), Musashi-1 (forward, 5′-GCAGACTACGCAGGAAGGG-3′; reverse, 5′-CGTGACAAACCCGAACCC-3′), and internal control GADPH (forward, 5′-TGAAGGTCGGAGTCAACGGA-3′; reverse, 5′-TGGTGCAGGAGGCATTGCTG-3′).
Biopanning in vitro
A Ph.D-12 peptide phage display library was purchased from New England Biolabs (Beijing, China). The kit used a combinatorial library of random peptide 12-mers fused to the N-terminus of a minor coat protein (PIII) of M13 phage. Negative/positive screen procedure was performed to obtain R-NS cells targeting phages, as shown in Figure 1B . For the first round of panning, a 10-µL phage library containing 1.5 × 1011 phages was added into 1-mL prepared R-ES cell suspension, as described above. The mixture was incubated at RT for 1 h with gentle agitation. The supernatant, containing R-ES cells unbinding phages, was recovered by centrifugation (3000 rpm, 3 min) and added to the prepared periphery cells (1 × 106) with gentle agitation for 1 h at RT. In the repeated centrifugation process, 1 µL of supernatant was collected for titering, and the remains were added to prepared R-NS cells (1 × 108) with gentle agitation for 90 min at RT. After washing 5 times with a washing buffer (PBS, 0.5% Tween-20), phages binding to the R-NS cells were eluted by a 1-mL elution buffer (0.1 M glycine-HCl, 0.1% BSA, pH 2.2) and neutralized with a 188-µL neutral buffer (1 M Tris-HCl, pH 9.1). The eluted phages were titered and amplified according to manufacturer’s instructions using the Escherichia coli strain ER2738. The procedure was performed 4 times. The binding efficiency of the phage pool was calculated by the ratio of its output number to the input number in the screening of each round.
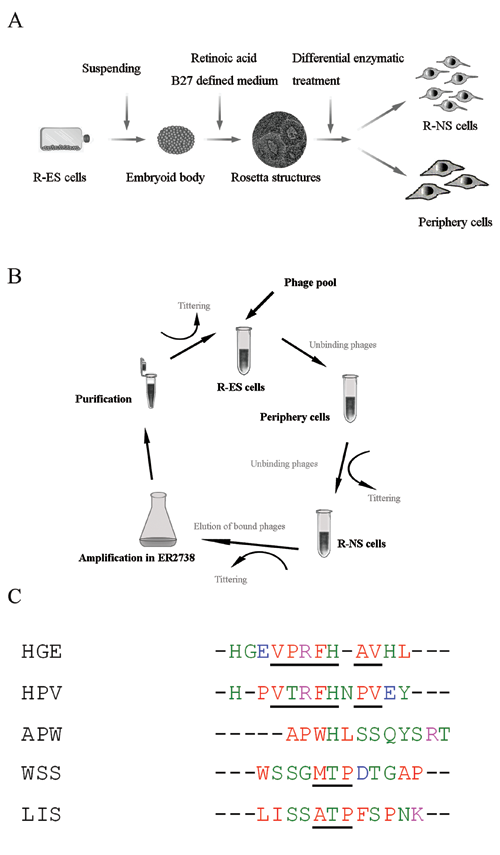
Biopanning in vitro. (
Sequencing of phage DNA, homology analysis, and database search
Phage plaques from the final round panning were amplified and purified as per the manufacturer’s instructions. The DNA of the selected monoclonal phages was sequenced by Invitrogen (Guangzhou, China). Multiple sequence alignments, clodogram creation, and identification of small peptide motifs adopted the CLUSTAL 2.0.10 multiple-sequence alignment (EMBL-EBI, http://www.ebi.ac.uk/Tools/clustalw2). The GenBank search adopted the NCBI protein-protein blast BLASTP 2.2.19. 12
Selection of targeting phages by whole-cell enzyme-linked immunosorbent assay
Confluent cells in a 96-well plate were fixed by 4% paraformaldehyde for 15 min at RT. After 3 washes (PBS) at 5 min each, a 250-µL blocking buffer (3% BSA-PBS) was added to each well, and the plate was incubated for 2 h at 37°C. Repeated 5-min washes (PBS) and 1 × 1010 phages in a 200-µL blocking buffer were added to each well and incubated for 90 min at RT with a gentle shake. Another set of repeated 5-min washes using 0.1% Tween-20-PBS was conducted and then rewashed 3 times with PBS, followed by incubation with horseradish peroxidase (HRP)–conjugated mouse anti-M13 antibody (1:1000 in a blocking buffer) for 90 min at RT. Repeated 5-min washes (PBS) were once again carried out, aspirated, and developed by 100 µL/well o-phenylenediamine for 20 min in the dark at RT, and stopped by 50 µL/well 2 M H2SO4. The supernatants were transferred in high-binding enzyme-linked immunosorbent assay (ELISA) plates (BBI, China) and were read on a GENios microplate reader (Tecan, Männedorf, Switzerland) at 492 nm. The absorbance at 405 nm was taken as a reference. Procedures were repeated 3 times.
Peptide synthesis
The selected peptide, HGEVPRFHAVHL, and the control peptide, VRAHEHLVGFPH, which contains the same peptide sequences as the HGE peptide but has different orders, were synthesized by a peptide synthesizer (CSBio 936, CSBio, Menlo Park, CA). Peptides were determined by mass spectrometric analysis (VarianProstar 210, Beckman Gold Systems, Beckman Coulter, Fullerton, CA).
Determination of binding efficiency and specificity by competition with free peptides
To analyze the binding ability of the selected phage to R-NS cells, the synthetic peptide HGEVPRFHAVHL was added to cells at various concentrations ranging from 1 nM to 10 µM before phages were added during the process of biopanning, as described above. Another peptide, SLHSQPRSWTAY, was used as a negative control. The binding ratio of phages to target cells was calculated by the output titer of phages selected with the competition of peptide normalized to the output titer of phages selected without adding peptides.
Immunocytochemistry of the selected peptide by QD-labeling
CdSe-ZnS core shell QDs (Evident Technologies, Troy, NY) with emission maxima centered at 580 nm were conjugated with HGE peptide via covalent bonding using EDC. The peptides were reacted with QDs bound to Sulfo-NHS for 2 h at RT and then stored at 4°C overnight.
QD-labeled peptides were incubated with cells to identify a specific binding ability of a selected sequence. For the immunocytochemistry step, cells were plated in a 1% gelatin-coated 24-well plate after adherence, based on the protocol described above. For the suspending step, cells were suspended in a blocking buffer (3% BSA in PBS) at 1 × 104 cells/mL and incubated for 30 min at 37°C. After they were washed twice with PBS, cells were mixed with 5 µL QD-labeled peptide and 100 µL 4% paraformaldehyde in a 400-µL blocking buffer with a gentle shake at RT for 1 h. They were washed 3 times with 0.1% Tween-20-PBS. Finally, they were counterstained with Hoechst 33258 (Invitrogen) for 15 min at RT and detected by an FV1000 Confocal laser scanning biological microscope (Olympus, Tokyo, Japan).
Western blot stained with the selected phage
Membrane proteins from the R-NS cells were extracted by a membrane extract kit (Keygentec, Nanjing, China) and were resolved on 12% sodium dodecyl sulfate (SDS)–polyacrylamide gels, then electrophoretically transferred to a PVDF membrane (Millipore, Billerica, MA). The blot was blocked with PBS with 3% BSA for 1 h and incubated with 3 × 109 phages in PBS with 3% BSA for another hour. After washing 3 times with 0.05% Tween-20-PBS, the membrane was incubated with an HRP conjugate mouse anti-M13 antibody (1:1000 dilution in PBS, 3% BSA) for 1 h. The membrane was further developed using BeyoECL Plus Kit (Beyotime, Haimen, China), and the blot was detected with ECL by exposing the membrane to an X-ray film (Kodak, Tokyo, Japan). Molecular weight was determined with a prestained protein molecular weight marker (Fermentas, Glen Burnie, MD).
Results and Discussion
The search of new receptor ligands is of great importance for the study of the ES cell differentiation process. The purpose of this study was to isolate novel peptide ligands that bind R-NS cells derived from rhesus monkey ES cells. The R-NS cells were obtained by retinoic acid treatment through the embryoid body formation and then isolated by differential enzymatic treatment ( Fig. 1A ). EB formation step was used to remove self-renewing signals (e.g., following separation from mouse embryonic fibroblasts) and to initiate cellular differentiation through cell-cell interactions. 13 After 6 days of EB formation ( Fig. 2A.a ), cells were continuously exposed to RA over the next 4 days following the formation of the EBs cultured in the NS cell–defined medium (B27) over 6 days, after which the central EB cells had generated rosette structures ( Fig. 2A.b ). There were clear edges between the rosetta structure and the periphery cells ( Fig. 2A.c ); immunocytochemical analyses demonstrated that the vast majority of rosette cells were positive for nestin (85%), SOX2 (88%), and Musashi-1 (95%), whereas the surrounding flat cells were negative ( Fig. 2A.d-f , 2.B ). A differential enzymatic treatment method was used to separate NS cells from periphery cells. In addition, the short-term adhesion method was carried out to remove contaminated single cells. 14 The isolated rosette cells were also positive for Musashi-1, nestin, and SOX2 ( Fig. 2A.g-i ), suggesting that the enzyme treatment method was valid to enrich ES cell–derived NS cells. Reverse transcription PCR was analyzed for the expression of markers characteristic of specific cell types, including ES cell marker OCT4, SOX2, neural precursor marker SOX2, nestin, and Musashi-1 ( Fig. 2C ). The expression of OCT4 occurred only in R-ESC cells, SOX2 both in R-ES cells and in R-NS cells, and nestin and Musashi-1 only in R-NS cells. A weak expression of Musashi-1 was detected in the periphery cells and d-NS cells (continuous cultured R-NS cells >40 days), indicating the expression of the key markers at different ESC differentiation stages.
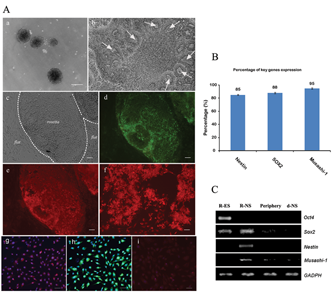
Enrichment and identification of the neural stem (NS) cells derived from rhesus monkey embryonic stem cells (R-ES cells). (
For the biopanning step, a procedure in liquid phase from suspension was performed, wherein the receptors were observed to more likely stay in their native conformation. 15 A negative/positive selection approach was employed, and the periphery cells and rhesus monkey ES cells were used as a negative selection ( Fig. 1B ). After 4 rounds of in vitro panning, phage pools exhibited a tendency of decreased binding efficiency with R-ES cells and periphery cells. Meanwhile, they showed an increasing recovery rate from 5.8% to 92% and an increasing binding efficiency with R-NS cells derived from R-ES cells, from 2.5 × 10−6 to 7.3 × 10−1 ( Table 1 ). Results indicate that the process of negative selection effectively reduced nonspecific binding phages to R-NS cells, but it successfully accumulated targeting phages. Phage yield rate was determined by calculating the ratio of output number to input number in each round of screening. Twenty-six monoclonal phages were picked and sequenced randomly. Among these phages, 11 showed the same sequence as APWHLSSQYSRT, 6 showed HGEVPRFHAVHL, 2 showed HPVTRFHNPVEY, and 2 single sequences showed WSSGMTPDTGAP and LISSATPFSPNK ( Fig. 1C ). The remaining 5 monoclonal peptide sequences were shorter than 12 amino acids. Although no notable homology was shown from these selected clones, potential consensus peptide motifs, such as V(P/T)RFH(A/P)V and (M/A)TP, appeared in these ligands ( Fig. 1C ) and are italicized.
Selection of Phage Antibodies against R-ES Cell-Derived Neural Stem Cells
Number of input phages.
Number of phages in supernatant before positive selection.
Number of phages contained in eluate.
Phage recovery rate = output phage/supernatant phage.
Phage yield rate = output phage/input phage.
Whole-cell ELISA results showed all phage candidates having high binding efficiency to R-NS cells, but only HGE (HGEVPRFHAVHL), WSS (WSSGMTPDTGAP), and HPV (HPVTRFHNPVEY) phages were observed to have comparatively low binding efficiency to both R-ES cells and periphery cells ( Fig. 3A ; p < 0.05). The APW (APWHLSSQYSRT) phage and LIS (LISSATPFSPNK) phage presented high binding efficiency to R-ES cells and periphery cells, respectively, suggesting that neither of them were appropriate candidates. In a further study, the binding affinities of HGE, HPV, and WSS (WSSGMTPDTGAP) phages with R-NS cells and differentiated R-NS cells (continuously cultured >40 days) were reanalyzed by whole-cell ELISA, with HGE phage displaying significant high binding efficiency to R-NS cells and low binding efficiency to differentiated R-NS cells ( Fig. 3B ; p < 0.01). This suggests that the HGE phage is the best candidate.
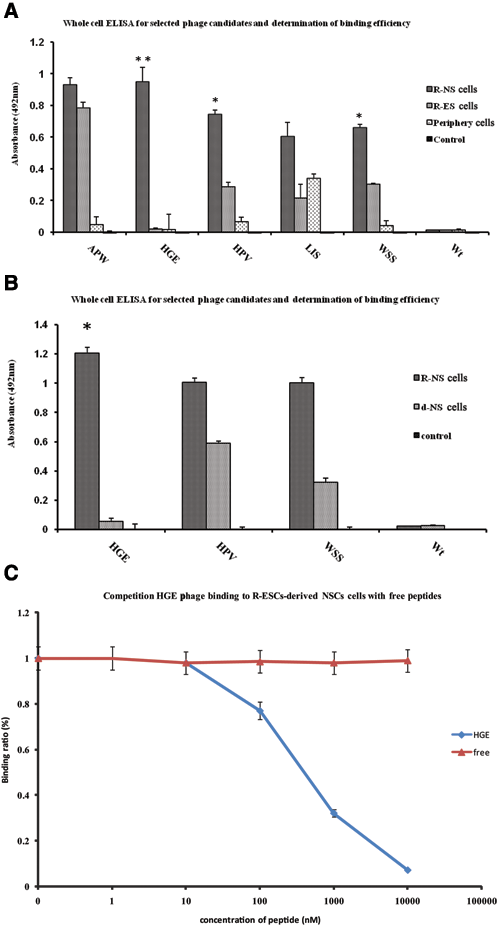
Whole-cell enzyme-linked immunosorbent assay (ELISA) results for the selected phage candidates and competition of the HGE phage binding to R-NS cells with free peptides. (
Following, a chemically synthetic peptide containing an HGEVPRFHAVHL sequence was used to test its competitiveness with the R-NS cells. Results show that this synthetic HGE peptide could compete with the HGE phage, achieving 50% inhibition at the concentration of about 500 nM ( Fig. 3C ). A control peptide with a sequence of VRAHEHLVGFPH failed to compete with the HGE phage for binding to NS cells, demonstrating that the binding ability of the phage to NS cells was caused by the displayed HGE peptide.
QD-labeled HGE peptides were incubated with R-ES cells, R-NS cells, periphery cells, and differentiated R-NS cells, whereas QD-labeled free peptides were incubated with R-NS cells as control. Strong fluorescence signals were shown specifically at the R-NS cell surfaces ( Fig. 4A.a ), whereas the control revealed only background staining and nonspecific fluorescent points ( Fig. 4A.b ). Meanwhile, the R-ES cells, periphery cells, and differentiated NS cells derived from R-ES cells did not show any obvious fluorescent specific signals ( Fig. 4A.c-e ). The same suspended step with the panning procedure was used here to monitor binding ligand on an R-NS cell membrane. As shown in Figure 4B.a-c , the cluster of fluorescence signals spread at the membrane of R-NS cells. Three-dimensional images were created by the overlap Z axis of 50 images at the different focus ( Fig. 4B.d ). The results confirmed that the HGE peptide binding to the R-NS cell’s surface with specificity and QD labeling extends the utilization of HGE peptide in this study.
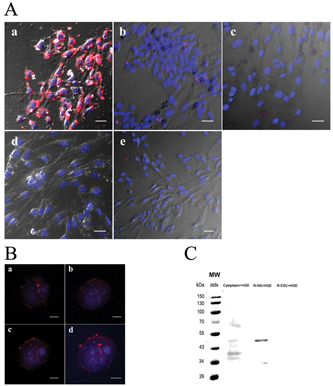
Immunofluorescence of QD-labeled HGE peptide and Western blotting of HGE phage. (
Western blot staining revealed doublet protein bands of about 48 and 34 kDa on membrane proteins of the R-NS cells, which correspond with the HGE peptide ( Fig. 4C ) at the third lane. Staining also revealed 5 bands from about 70 to 36 kDa on the cytoplasmic proteins of the R-NS cells, at the second lane, suggesting HGE phage also may have had protein-protein interactions with cytoplasm proteins. Compared with a control sample stain of membrane proteins of R-ES cells at the fourth lane, no obvious band could be observed.
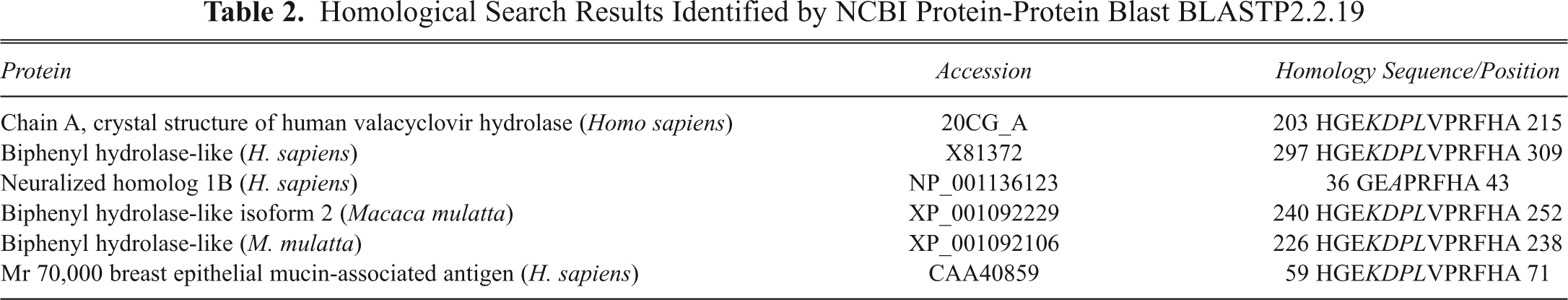
Because peptides obtained by biopanning procedures often possess the unique property of binding to molecules involved in important functional interactions, 8,16 the active sites of receptors are frequently based on the high degree of homology between the peptide and ligand. 17,18 A GenBank protein blast search revealed several relative proteins of Homo sapiens and Macaca mulatta ( Table 2 ), and 2 proteins, known as neutralized homolog 1B and breast epithelial mucin-associated antigen, are of particular interest. Neutralized homolog 1B was reported to regulate a Notch ligand in cooperation with Mind bomb-1 and the breast epithelial mucin-associated antigen. 19 Notch pathway molecules are essential in the maintenance of mammalian NS cells, 20 and the breast epithelial mucin-associated antigen has been the last search result shown in Table 2 . Although the overall significance of the HGE peptide is unknown, we believe that the homological sequence HGE—VPRFHA, as shown by the blast search, sharing a consensus sequence of VPRFHA with the V (P/T) RFH (A/P) V motif, may be important in the binding of individual phage clones to the NS cell surface. Meanwhile, the receptor ligands may have some relationship with the Notch pathway. Work should be conducted further to assess the selected sequence.
Homological Search Results Identified by NCBI Protein-Protein Blast BLASTP2.2.19
Our present work provides an essential clue for the selection of receptor ligands vis-à-vis fixed cell populations from ES cell differentiation. We show that the peptide obtained from biopanning can be conjugated with fluorescence dyes and can effectively label R-NS cells. The study also determined the corresponding proteins on the cell surface, revealing the underlying pathways of specific cell lineages. The techniques used in this study could be applied flexibly to other cell lineages derived from ES cells.
Footnotes
Acknowledgements
Financial support by Hi-tech Research and Development Program of China (No. 2006AA03Z359) is gratefully acknowledged. Special thanks to Ni Hu, Wugen Zhan, and Chuang Kou for technical assistance. We are also thankful to Jing Ge and Professor Lijun Wang for the critical reading of the manuscript.