
Abstract
Dietary long-chain fatty acid (LCFA) uptake across cell membranes is mediated principally by fatty acid transport proteins (FATPs). Six subtypes of this transporter are differentially expressed throughout the human and rodent body. To facilitate drugs discovery against FATP subtypes, the authors used mammalian cell lines stably expressing the recombinant human FATP4 and 5 and developed a high-throughput screening (HTS) assay using a 96-well fluorometric imaging plate reader (FLIPR). LCFA uptake signal-to-background ratios were between 3- and 5-fold. Two 4-aryl-dihydropyrimidinones, j3 and j5, produced inhibition of FATP4 with a half-maximal inhibitory concentration (IC50) of 0.21 and 0.63 µM, respectively, and displayed approximately 100-fold selectivity over FATP5. The US Drug Collection library was screened against the FATP5. A hit rate of around 0.4% was observed with a Z′ factor of 0.6 ± 0.2. Two confirmed hits are bile acids, chenodiol and ursodiol with an IC50 of 2.4 and 0.22 µM, respectively. To increase throughput, a single time point measurement in a 384-well format was developed using the Analyst HT, and the results are comparable with the 96-well format. In conclusion, the FATP4 and 5 cell-based fluorescence assays are suitable for a primary drug screen, whereas differentiated cell lines are useful for a secondary drug screen.
Keywords
Introduction
L
Growing evidence indicates that LCFA uptake is tightly regulated by a number of plasma membrane–associated proteins, including fatty acid translocase, plasma membrane–bound fatty acid binding protein, and long-chain fatty acyl-CoA synthetase. 4,5,13-15 Furthermore, hormonal regulation of FATP activity may play an important role in energy homeostasis. 16 In adipocytes, the adipogenic hormone insulin increases expression of the long-chain fatty acyl-CoA synthetase 17 and induces plasma membrane translocation of FATPs from an intracellular perinuclear compartment to the plasma membrane. This process is paralleled by an increase in LCFA uptake. 16 Chronic leptin administration decreases fatty acid uptake, 18 and acute leptin application has a direct inhibitory effect on insulin-stimulated fatty acid uptake. Furthermore, treatment with tumor necrosis factor–alpha inhibited basal- and insulin-induced LCFA uptake and reduced FATP1 and FATP4 expression levels. 16 Disturbed fatty acid metabolism and homeostasis are associated with insulin resistance. In obesity and other metabolic syndromes, dysfunction of these FATPs may contribute to elevations in serum-free fatty acids and the genesis of type 2 diabetes. 4,19,20
Lacking biologic probes for each subtype of the FATP family, studies of the physiologic role of an individual subtype principally have relied on gene knockout mouse models in vivo and overexpression or antisense knockdown in vitro. 4,12,20-22 FATP1 knockout mice displayed less insulin resistance following lipid infusion or a high-fat diet compared to wild-type mice. 20 In contrast, homozygote deletion of the FATP4 gene led to early embryonic lethality. Although this result illustrates an essential role for FATP4 in embryonic development and survival, the knockout model failed to give insight into a potential role for FATP4 in lipid absorption or insulin resistance. 12,22 Nevertheless, it was speculated that a selective inhibitor against FATP4 could specifically reduce LCFA absorption with minimal interfering with other nutrient intakes. 4 FATP5 is exclusively expressed in the liver and localized to the basal plasma membrane of hepatocytes, congruent with a role in LCFA uptake from the circulation. Overexpression of FATP5 in mammalian cells increased the uptake of 14C-oleate and fluorescence-labeled LCFA. 4 Conversely, LCFA uptake was reduced in vitro in hepatocytes isolated from FATP5 knockout mice. The FATP5 knockout mice have lower hepatic triglyceride and free fatty acid content in the liver compared to wild-type mice. 23 Furthermore, detailed phenotypic analysis unexpectedly revealed that FATP5 deletion mice did not become overweight under a high-fat diet. Because FATP5 does not express in the gastrointestinal tract, FATP5 null mice had normal fat absorption. This phenotype likely resulted from both a decrease in general food intake and an increase in energy expenditure. 23 These findings not only indicate an important role for FATP5 in the regulation of liver fat content but also suggest FATP5 possibly being involved in body weight homeostasis. Therefore, a selective and potent FATP5 inhibitor could provide a powerful biologic probe to further study the FATP5 function. Furthermore, it could provide a basis for the development of a specific therapeutic for treatment of fatty liver disease, where abnormal fat metabolism is primarily responsible for pathogenesis.
High-throughput screening (HTS) of a large compound library against an FATP subtype could be the initial step toward identifying novel subtype-selective compounds that specifically inhibit LCFA absorption or transport. Conventional methods for the assessment of LCFA uptake using radiolabeled fatty acids and fluorescent fatty acid analogs with fluorescence-activated cell sorters (FACS) are very slow and not suitable for HTS in drug discovery. Recently, an LCFA uptake measurement method using the QBT Fatty Acid Uptake Assay Kit (QBT kit) has been used to determine real-time LCFA uptake kinetics in 3T3-L1 adipocytes using Gemini or Flexsation fluorescence plate readers. 24,25 The 3T3-L1 adipocytes require differentiation from fibroblast-like progenitors, a process that has been inconsistent and time-consuming. Moreover, 3T3-L1 adipocytes are murine in origin and express several proteins involved in LCFA uptake in addition to FATP1 and FATP4. 14,26 Thus, a defined target inhibitor is not easily identifiable. To overcome this problem, HTS assays using live yeast cells expressing murine or human FATP2 have been developed and used either a 96- or 384-well format to identify small compounds that reduced the import of a fluorescent fatty acid analog, 4,4-difluoro-5-methyl-4-bora-3a,4a-diaza-s-indacene- 3-dodecanoic acid (C(1)-BODIPY-C(12)) by FATP2. 27,28 In the present study, we have developed an HTS assay (96- or 384-well format) against recombinant hsFAPT4 or FATP5 stably expressed in mammalian cell lines for the screening of subtype-specific inhibitors using a fluorometric imaging plate reader (FLIPR) or the Analyst HT with the QBT assay reagent.
Materials and Methods
HsFATP4 and 5 stably expressing cell lines and reagents
Creation of human embryonic kidney (HEK) 293 cell lines stably expressing human recombinant HsFATP1, 4, and 5 has been described previously. 4 Cell culture media and supplements were purchased from Gibco Invitrogen (Invitrogen, Carlsbad, CA). The QBT Fatty Acid Assay Kits were purchased from MDS Analytical Technologies (formerly Molecular Devices Co., Sunnyvale, CA). Chenodeoxycholic acid, oleic acid, and palmitic acid were purchased from Sigma-Aldrich (St. Louis, MO). The 2 FATP4 inhibitors, j3 and j5, were synthesized based on a recent publication. 29 A US Drug Collection (USDC) library consisting of 1040 compounds with known pharmacology was purchased from Microsource (Gaylordsville, CT). All compounds were dissolved in pure DMSO at 10 mM as stock solutions.
Cell cultures
The hsFATP1, hsFATP4, or hsFATP5 cells and vector control HEK293 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 2250 mg/L D-glucose, pyroxidine hydrochloride, and 0.5 mM CaCl2; supplemented with 10% fetal bovine serum (FBS) and 100 units penicillin–100 µg streptomycin/mL; and supplemented with 1 mg/mL G418 (for genetic selection). Cells were seeded into PDL-coated black-walled, clear-bottom microplates (Becton Dickinson, Franklin Lakes, NJ) at a cell density of 80,000 cells/well for the 96-well format and 20,000/well for the 384-well format. Plates were used for assay 12 to 16 h after seeding.
3T3-L1 preadipocytes (American Type Culture Collection, No. CCL92.1) were differentiated following the published protocol. 24,25 Cell differentiation was assessed by visual inspection of lipid droplet accumulation under a microscope, and cells were seeded into 96-well plates at 100,000 cells/well 12 to 16 h before the assay.
QBT kit and fluorescence intensity measurement
The QBT mixture was prepared as instructed in the product insert (R8133, MDS Analytical Technologies) and consisted of a proprietary quenching agent and 2 µM of the fluorescently labeled fatty acid BODIPY-FA. The cell-impermeable quenching agent effectively quenched the fluorescence of BODIPY-FA in solution. Cells took up the BODIPY-FA, and intracellular fluorescence was measured using a bottom-read detector FLIPR or Analyst HT (both from MDS Analytical Technologies). All synthetic small molecules were dissolved in DMSO as stock solutions and then diluted with 1× loading buffer (1× Hank’s balanced salt solution with 20 mM HEPES buffer and 0.2% fatty acid free bovine serum albumin [BSA]). For kinetic assays in the 96-well format, culture media in the plate were replaced with 150 µL 1× loading buffer plus 50 µL diluted compounds at 5× concentration (or 175 µL 1× loading buffer plus 25 µL diluted compounds at 10× concentration) and incubated for 30 min at 37°C in a 5% CO2 incubator. At the end of the incubation time, 50 µL of the QBT mixture was added to the well. To examine the inhibitory effect of fatty acid derivatives, we dissolved them in ethanol as stock solutions and diluted them with 1× loading buffer. The dilution was then equilibrated with the QBT mix for 24 h in 4°C before being added to the plate for the uptake assay. Immediately after the addition of QBT mix to cells, fluorescence readings were started using the FLIPR with an excitation filter of λex = 488 nm and an emission filter of λem = 540 nm at room temperature. For single time point assays in the 384-well format, culture media in the plate were replaced with 35 µL 1× loading buffer plus 5 µL diluted compounds at 10× concentration and incubated for 30 min at 37°C in a 5% CO2 incubator. At the end of the incubation, 10 µL of the QBT mixture was added to the well. Then, 1 min and 1 h following the dye incubation, fluorescence intensity of each well was measured using the Analyst HT with an excitation filter of λex = 488 nm and an emission filter of λem = 540 nm at room temperature. The pilot drug screens were conducted in a blinded manner, and the identities of the hits were not decoded until the completion of the verification using concentration-response tests.
Data analysis
For kinetic assays, the uptake of BODIPY-FA was observed continuously for 1 h on the FLIPR. The relative fluorescence unit (RFU) level of each well was normalized by subtracting the fluorescence level in the first data point. The endpoint RFU after the 1-h observation was used as the maximum fatty acid uptake. For the single time point assay in the 384-well format, the absolute fluorescence intensity of each well was obtained as the maximum fatty acid uptake. Percentage changes in fluorescence signals (inhibition or enhancement) compared with experimental controls were calculated from the mean of duplicate measurements in the primary assay development. Because we cannot measure the final free fatty acid concentrations in the wells, the potency of inhibition by fatty acids was based on the starting concentrations with the assumption that they bind to proteins in the growth medium to a similar degree. Similarly, the small-molecule potency was estimated based on their apparent molar concentrations because we did not determine their protein binding profiles. Concentration-response curves were constructed and fitted to a 4-parameter sigmoidal function to yield a half-maximal inhibitory concentration (IC50). As appropriate, data were represented as mean ± SEM of at least 3 measurements.
The Z′ factor reflects both the assay signal dynamic range and the control data variation associated with the signal measurement. 30 The Z′ factors from each plate were calculated based on the following equation:
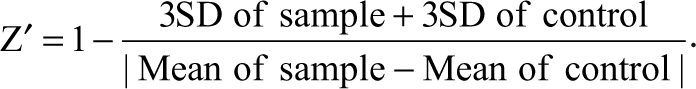
To identify initial hits (inhibitory compounds) from the pilot screen at a single measurement, we used the statistical z score—that is, it is the ratio of the difference between the sample value (X) and the mean of all wells in the entire plate over the standard deviation (SD) of the mean. In this case, the z score was calculated using the following formula:

The inhibitory compounds were defined in our pilot screen with a z score ≤ –3 z (i.e., with 3 SD from the mean of compounds in the entire plate).
Results
Signal-to-background ratio for the assay using hsFATPs-expressing cells
We started with determining the signal-to-background ratio by comparing active LCFA uptake in hsFATP1-, 4-, or 5-expressing cells to vector control cells (HEK293 cells transfected with an empty expression vector and selected under the same conditions as FATP-expressing cells) to evaluate the specific signal over nonspecific uptakes for the HTS assay. As
Kinetic signals of fluorescently labeled long-chain fatty acid (BODIPY-FA) uptake in 4 stable cell lines expressing recombinant hsFATP1, FATP4, FATP5, and FATPc (vector control) using a fluorometric imaging plate reader (FLIPR). (
Competitive inhibition of BODIPY-FA uptake by fatty acid derivatives
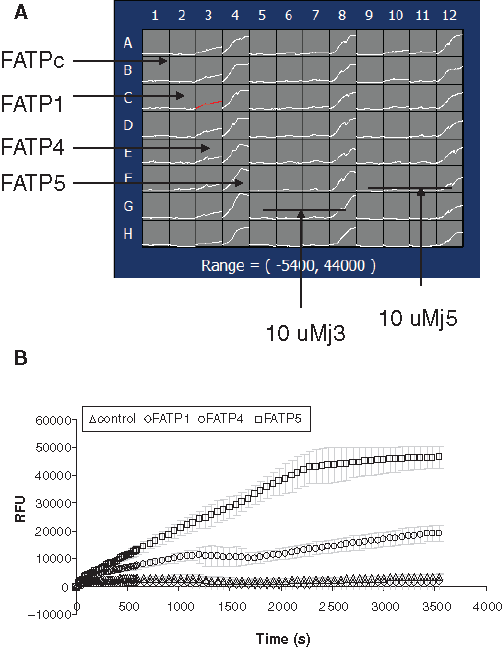
The fluorescently labeled fatty acid BODIPY-FA is actively transported through the cell membrane by the FATPs. Therefore, unlabeled fatty acid derivatives should act as competitive inhibitors to decrease the intracellular fluorescence. Natural substrate oleic acid competitively inhibited the FATP4 uptake with an IC50 of approximately 3.1 µM with 2 µM BODIPY-FA in the QBT mix solution (
Inhibition of fluorescently labeled BODIPY-FA uptake by natural long-chain fatty acids in the hsFATP4-expressing cells. (
Selective inhibition of FATP4 by 4-aryl-dihydropyrimidinones
During development of the HTS assay, a report described, for the first time, a class of small molecules (4-aryl-dihydropyrimidinones) that produce potent inhibition of hsFATP4.
29
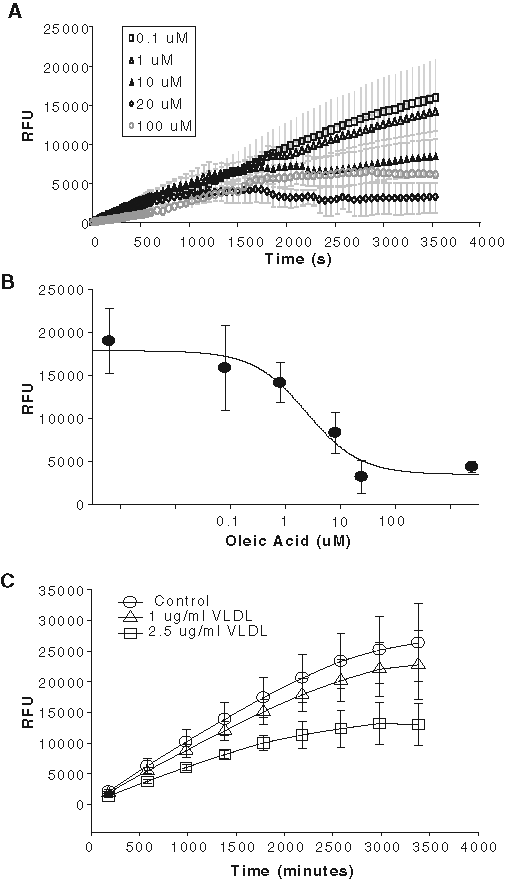
Two selected compounds in the published chemical class, j3 and j5, were synthesized and tested. As shown in
Two small molecules, j3 and j5, selectively inhibit fatty acid uptake through FATP4 over FATP5. (
The highest concentration of DMSO contained in the j3 and j5 testing wells was 0.1%. Because in primary screening, most compounds are dissolved in DMSO as stock solutions, we examined assay tolerance to this most commonly used solvent. Varying concentrations of DMSO (0.05%-1%) were added to hsFATP4 or 5 cells, vector control cells, and 3T3 L1 adipocytes and incubated for 30 min at 37°C prior to LCFA uptake measurement by the FLIPR. DMSO at ≤1% at the preincubation time used here apparently did not cause significant signal changes in hsFATP4, hsFATP5, and 3T3L-1 cells (
Pilot drug screen against hsFATP5
We further evaluated the quality and reproducibility of the assay by conducting a pilot screening of the USDC library consisting of 1040 compounds. By the time we developed this assay, selective FATP4 inhibitors (such as j3 and j5) had just become available,
29
but no FATP5 specific inhibitors or HTS assay against FATP5 target have been reported thus far. We therefore concentrated on our initial screening against hsFATP5. All test compounds were dissolved in DMSO at 10 mM as stock solutions and screened at a final single concentration of 10 µM in a single well measurement. For each plate, the identification of active compounds (hits) was based on the z score of each compound as described in Materials and Methods. Compounds with z scores ≤ –3 indicate potential inhibiting, whereas scores ≥ +3 indicate enhancement of BODIPY-FA uptake. Using this selection criterion, 9 hits of 1040 were identified. In the following concentration-response tests using the same assay in a duplicated measurement, 4 inhibitory compounds were verified among the initial hits, indicating a hit rate at 0.4% for the USDC library. Two of the confirmed hits were bile acids, chenodiol (chenodeoxycholic acid) and ursodiol (ursodeoxycholic acid).
31
Further tests revealed that chenodiol and ursodiol caused concentration-dependent inhibition of hsFATP5 with an IC50 of 2.4 µM and 0.22 µM, respectively (
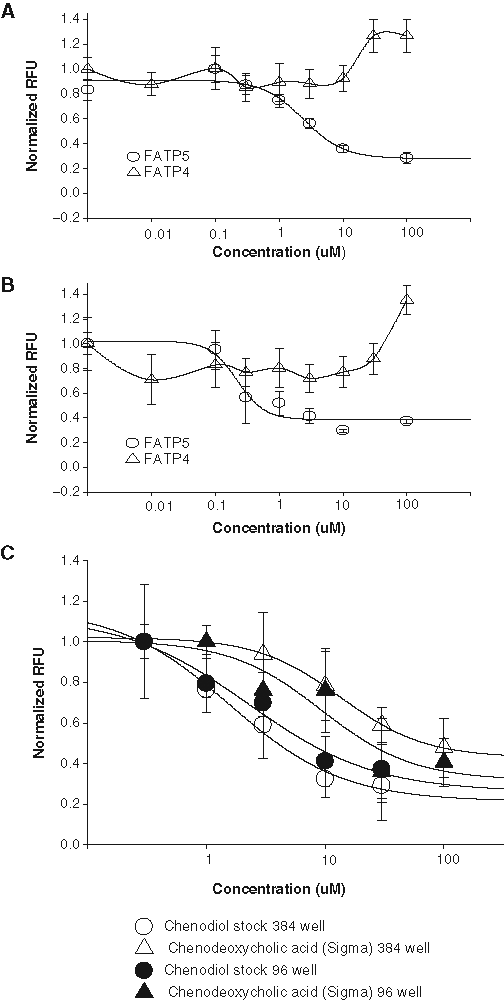
(
Single time point fluorescence intensity measurement in a 384-well plate format
To demonstrate that this fluorescence-based hsFATP-cell assay can be run in an HTS format, the assay was modified into a 384-well format with a single time point measurement using a conventional plate reader (Analyst HT). Cells expressing hsFATP5 were plated in 20,000/well 12 to 16 h before the assay. After loading of dye, the fluorescence intensity of each well was measured immediately (as the baseline) and at a 1-h time point following dye incubation. Seven plates were tested with Z′ = 0.6 ± 0.1, similar to that observed in a 96-well assay. The IC50 of chenodiol (2.3 µM) obtained in the 384-well assay is consistent with the value (1.6-2.4 µM) obtained from the 96-well assay. Chenodeoxycholic acid (Sigma) is the same chemical as chenodiol (US Drug Collection). The IC50 of chenodeoxycholic acid from Sigma obtained in the 384-well 13.5 µM is also similar to its potency (9.3 µM) determined by a FLIPR in the 96-well assay. These results indicate that the single time point measurement in the 384-well format is suitable for primary HTS.
LCFA uptake assay in 3T3 L1 adipocytes
LCFA uptake by native cells is likely to be more complex than by a recombinant cloned FATP expressed in a mammalian cell line. Moreover, this process is hormone regulated. We therefore compared stimulation and inhibition of fatty acid uptake by cells endogenously expressing FATPs. To this end, we started with 3T3 L1 adipocytes, which have been shown to express both FATP1 and 4. Differentiation protocol for 3T3-L1 cells followed the published procedure,
24,25
and the differentiated cells were assessed by visual inspection of lipid droplet accumulation under a microscope. 3T3-L1 adipocytes or undifferentiated fibroblasts were seeded into 96-well plates at 100,000 cells/well and treated with insulin (0.2-1.7 µM) as indicated in
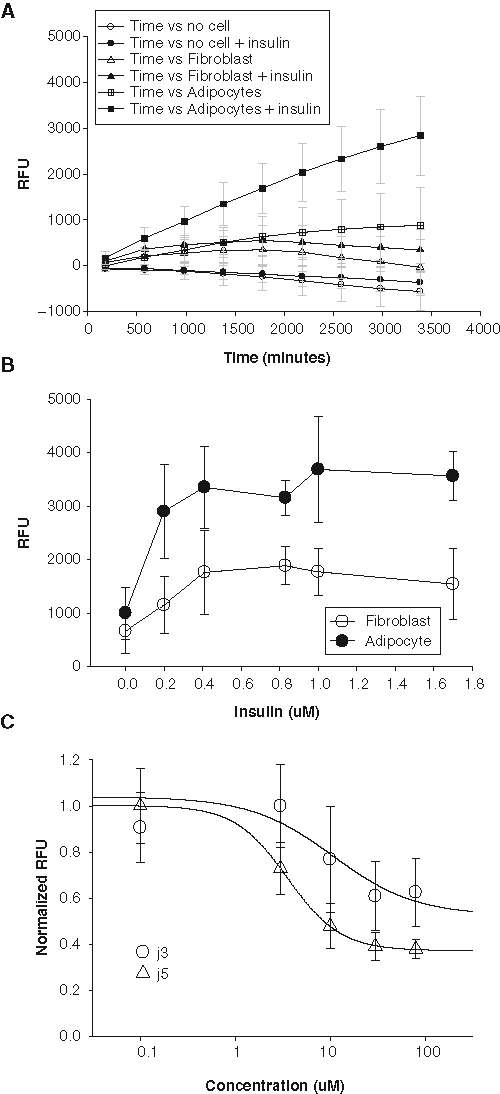
Fatty acid dye uptake in differentiated 3T3L1 adipocytes. (
Inhibition of LCFA uptake by FATP4 inhibitors in mouse enterocytes
FATP4 is the primary intestinal LCFA transporter and should be inhibited by the FATP4 inhibitors, such as j5. Thus, it is physiologically relevant to determine the effect of j5 on LCFA uptake by isolated primary mouse enterocytes. Using primary enterocytes for the FLIPR assay would require a large number of cells and will make it time-consuming and costly. Moreover, the nonadherent nature of enterocytes would affect the consistency of the assay. As an alternative, we used a flow cytometer-based assay in conjunction with BODIPY–fatty acid to measure short-term LCFA uptake following isolation of duodenal enterocytes. Enterocytes were exposed for 60 s to a 2-µM BODIPY-FA solution containing several different concentrations of j5 as indicated in
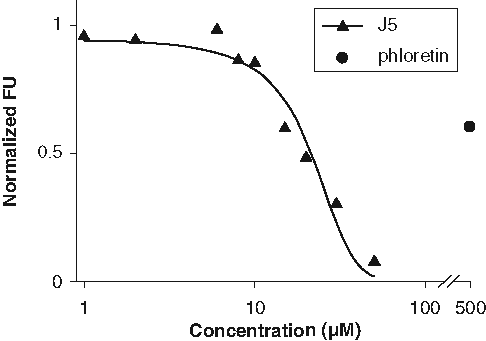
Fluorescence-activated cell sorter (FACS)–based long-chain fatty acid (LCFA) uptake assay using acutely dissociated mouse enterocytes. Cells were incubated with j5 (1-30 µM) or phloretin (500 µM), a nonspecific fatty acid transport protein (FATP) inhibitor. FACS was used to gate on viable cells and determine LCFA uptake expressed as percent of control (containing 0.5% DMSO). Curve represents a nonlinear regression (R 2 = 0.96) with an estimated IC50 of 21 µM.
Discussion
HTS assay viability using the stable cell lines expressing recombinant hsFATPs
Our study shows that the hsFATP4 and hsFATP5 cell-based fluorescence assays using a FLIPR or a conventional fluorescence plate reader are suitable for primary drug screening. Cellular LCFA uptake involves both passive diffusion and FATP-mediated transport. The background of fluorescence fatty acid uptake could be due to the passive diffusion or transportation by endogenous FATPs in HEK293 cells. The established hsFATP4 and hsFATP5 stable cell lines have substantial and significant signal-to-background ratio compared to the vector control cell line (
In the pilot screen using a small compound library of approximately 1000 (USDC) against the hsFATP5 to evaluate assay reproducibility, a Z′ factor ranging from 0.5 to 0.7 was obtained, indicating good assay reproducibility. Interestingly, 2 apparent hsFATP5 inhibitors identified in random screening of the USDC library were bile acids (
Our assay targeted 2 human fatty acid transporters, hsFATP4 and hsFATP5, expressed in a human mammalian cell line. Our study differs from the studies published by Li et al, 27,28 where a different fluorescent quencher, Trypan blue, and C1-BODIPY-C12 were combined to develop a yeast cell-based assay against murine or human FATP2. This assay relies on the functional expression of mammalian FATPs in a yeast strain lacking the endogenous fatty acid transporter. Although the use of yeast cells allows for rapid cell proliferation, it has the disadvantage of reconstituting the function of a mammalian transporter. Indeed, half of the 6 mammalian FATPs, including FATP5, do not enhance C1-BODIPY-C12 uptake when expressed in yeast, but these proteins are clearly able to enhance free fatty acid uptake when expressed in mammalian cells. The HEK cell-based assay presented here can be more easily adopted into typical primary drug screening pipelines along with other mammalian cells expressing different receptors or ion channels using common instruments such as a FLIPR 32 or a conventional plate reader (e.g., Analyst HT).
Differential effects of FATP4 inhibitors and bile acids against hsFATP4 and hsFATP5
The 2 confirmed hits are chenodiol (i.e., chenodeoxycholic acid), a primary bile acid formed by synthesis in the liver, and ursodiol (also known as ursodeoxycholic acid), one of the secondary bile acids, which are metabolic by-products of intestinal bacteria. Bile acids are known as substrates of FATP5 in the liver. FATP5 not only has been shown to be able to transport fatty acid through the cell membrane but also possesses bile acid–CoA ligase activity. 21 It is unclear whether the bile acids act as inhibitors directly on the transport protein or as substrates for bile acid–CoA ligase and indirectly inhibiting the uptake. The 2 bile acids have no inhibitory effect on the fatty acid uptake through FATP4, indicating that the bile acids cannot bind to the hsFATP4.
Secondary screens using native cells expressing FATPs
It is important to verify the hits identified from primary screening using recombinant cell lines with native cells that endogenously express the transporter of interests. 3T3-L1 adipocytes are murine in origin and express several proteins involved in LCFA uptake, with FATP1 and FATP4 as the predominant subtypes. 25 The reduced potency of the 2 FATP4 inhibitors, j3 and j5, in 3T3-L1 adipocytes compared to the recombinant hsFATP4 cell line suggests that the compounds may also have higher selectivity with FATP4 than FATP1. However, we also found a reduced efficiency of j5 in murine enterocytes, which do not express FATP1. This may indicate that in primary cell FATPs, the compound may interact with other accessory proteins such as CD36. Alternatively, the accessibility of the compounds to the native FATP proteins is limited. Unlike primary enterocytes, 3T3-L1 adipocytes could be used for secondary screening with a FLIPR. However, we found that the differentiation process was not only time-consuming (8-12 days from initiation of differentiation to the assay) but also inconsistent, yielding great variability in signal-to-background ratios among different batches of differentiated cells. Other native cell lines such as Caco-2 and HepG2 cells and 3 endothelial cell lines (b-END3, HAEC, and HMEC) express different FATPs that could potentially be used in secondary screening. 28,33
Potential drug classes acting as inhibitors of FATPs
A family of tricyclic, phenothiazine-derived drugs was shown to inhibit the hsFATP2 expressed in yeast, and such effects were linked to the possible cause of metabolic side effects, including hypertriglyceridemia in patients chronically taking this class of antipsychotic drugs. 28 Twenty-two compounds are labeled as “antipsychotic” drugs in the USDC library. In our pilot primary screening, these drugs did not produce any inhibition on hsFATP5, suggesting these antipsychotic drugs may have selectivity against FATP2 over FATP5 or result from the different expression systems. In contrast, our finding for the first time shows that bile acids cause concentration-dependent inhibition of LCFA uptake by FATP5 with apparent selectivity over FATP4. Bile acids have long been viewed as digestive detergents to facilitate fat absorption in the small intestine and the primary role in regulation of cholesterol catabolism. However, bile acids are now recognized as hormones involved in the regulation of various metabolic processes. 31 Our preliminary finding that bile acids can selectively inhibit the liver-specific FATP5, without effects on the intestine-predominant FATP4, further supports the notion of bile acids functioning as hormones and might have clinical implications. In conclusion, we have developed an HTS assay employing fluorescent fatty acid uptake by the recombinant human FATP4 and FATP5 stably expressed in HEK cells. This assay can be used for primary HTS for discovering novel compounds against hsFATP4 or hsFATP5, whereas the 3T3-L1 adipocytes or primary cells (e.g., enterocytes or hepatocytes) could be suitable in secondary screening for hit confirmation, lead identification, and optimization.
Footnotes
Acknowledgements
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke and the National Institute of Mental Health. We are grateful to Dr. Ling Jong for synthesis of compounds j3 and j5 for our research.
This work was supported by NIH R21NS57052 (Xie) and R01 MH078194 (Xie).