
Abstract
Huntington’s disease (HD) is associated with increased expression levels and activity of tissue transglutaminase (TG2), an enzyme primarily known for its cross-linking of proteins. To validate TG2 as a therapeutic target for HD in transgenic models and for eventual clinical development, a selective and brain-permeable inhibitor is required. Here, a comprehensive profiling platform of biochemical and cellular assays is presented which has been established to evaluate the potency, cellular efficacy, subtype selectivity and the mechanism-of-action of known and novel TG2 inhibitors. Several classes of inhibitors have been characterized including: the commonly used pseudo-substrate inhibitors, cystamine and putrescine (which are generally nonspecific for TG2 and therefore not practical for drug development), the various peptidic inhibitors that target the active site cysteine residue (which display excellent selectivity but in general have poor cellular activity), and the allosteric reversible small-molecule hydrazides (which show poor selectivity and a lack of cellular activity and could not be improved despite considerable medicinal chemistry efforts). In addition, a set of inhibitors identified from a collection of pharmacologically active compounds was found to be unselective for TG2. Moreover, inhibition at the guanosine triphosphate binding site has been examined, but apart from guanine nucleotides, no such inhibitors have been identified. In addition, the promising pharmacological profile of a TG2 inhibitor is presented which is currently in lead optimization to be developed as a tool compound.
Keywords
Introduction
T
To enable the discovery and characterization of potent and selective TG2 inhibitors, we have developed and optimized a large set of biochemical and cellular assays that are requisite for compound advancement. The primary biochemical and cellular assays are directed toward the transamidation activity of the target TG2 itself. Secondary assays have been designed to measure selectivity among the TG isoforms as well as species, cross-reactivity to other catalytic cysteine-containing enzymes (caspase-3), and other side effects such as cytotoxicity. In addition, to identify the binding site and mode of action for these inhibitors, mechanism-of-action assays have been developed.
The selectivity profile across other transamidating enzymes is an important factor in the development of TG2 inhibitors for both target validation and therapeutic applications. Among the large family of transglutaminase enzymes, we have established secondary biochemical assays for recombinant TG1, TG3, TG6, and factor XIIIa enzymes. Inhibition of any of these subtypes could contribute to detrimental side effects, such as ichthyosis for TG1 activity 14 and compromised blood clotting with factor XIIIa inhibition. 15 In addition, because all transglutaminases contain an active site cysteine residue that is prone to be the reactive moiety for irreversible inhibitors, we have also established a caspase-3 assay, an enzyme that also contains a reactive cysteine in its active site, as a counterassay to assess promiscuous nonspecific thiol-reacting compounds. Last, we have established a mouse TG2 biochemical assay to assess ortholog activity because the first in vivo testing of compounds will be in mouse models of HD.
The assays described here have been used to characterize published inhibitors of TG2—namely, a set of irreversible peptide-like inhibitors, the often used cystamine compound, 16 the reported quinazolinone hydrazide scaffold, 17 a set of inhibitors from the LOPAC and Prestwick collection of drugs, 18 and inhibition via the guanosine triphosphate (GTP) binding site have been characterized using guanine nucleotides. Each of these compound sets was found to be intractable for in vivo testing because of a general lack of selectivity or poor cellular potency. In parallel to using the profiling platform for known inhibitors, we present a promising profile of a novel inhibitor that is currently being developed and optimized for eventual testing in HD mouse models and potential therapeutic development.
Materials and Methods
Transglutaminase reagents
Transglutaminase enzymes, including human TG1, TG2, TG3, TG6, factor XIII, and mouse TG2, were purchased from Zedira (Darmstadt, Germany).
Biochemical reagents
The dansyl-labeled amine nucleophile (KxD) was synthesized using commercial reagents, Boc-Lys-OH (Fluka, St. Louis, MO), and dansyl-ethylendiamine (Invitrogen, Carlsbad, CA), and to avoid the formation of by-products, protection of the side chain amine of Boc-Lys-OH was performed using the selectively cleavable 4-methoxytrityl (Mtt) protecting group. Subsequently, the amine-protected lysine was activated and conjugated with dansyl-ethylendiamine. Finally, the Mtt protecting group was cleaved by mild acidolysis, and the resulting product mixture was purified by reversed-phase liquid chromatography (RP-HPLC).
Compounds
Cystamine and guanosine analogs were purchased from Sigma-Aldrich (St. Louis, MO). Z-DON and Boc-DON were purchased from Zedira. Other inhibitors were synthesized at Evotec (Hamburg, Germany) according to the published methods, including the irreversible peptide inhibitors CHDI-00314996, CHDI-00315191, and CHDI-00315813 19 ; the irreversible peptide inhibitors CHDI-00312412, CHDI-315216, and CHDI-313915 20 ; the irreversible dipeptide inhibitors 21 ; the reversible inhibitors CHDI-00194506 and CHDI-00194507 22 ; and CHDI-00209424 and CHDI-00208889 (analogs prepared by Evotec as part of SAR exploration of CHDI-00194506 and CHDI-00194507). The compounds reported by Lai et al. 18 were purchased from the following sources: tyrphostin 47, N-1786, ZM 449829, ZM 39923, GW 5074, and Rottlerin from Tocris Bioscience (Ellisville, MO); Menadione from Alfa Aesar (Ward Hill, MA); and β-lapachone from BioMol International (Plymouth Meeting, PA).
Fluorescent transamidation assays
The fluorescent screening assay for human TG2 was performed as described previously. 22 Assay conditions were 20 nM TG2, 8 µM N,N-dimethylated casein (NMC), and 16 µM KxD (used for all transglutaminase assays) in 25 mM HEPES (pH 7.4), 250 mM NaCl, 2 mM MgCl2, 0.5 mM CaCl2, 0.2 mM dithiothreitol (DTT), an 0.05% Pluronic F-127 at 37 °C. A kinetic measurement was recorded (Safire or Ultra, Tecan, Foster City, CA; ex: 350 nm, em: 535 nm), and the reaction velocity was used as a measure for enzyme activity. Assay conditions were identical for human TG6 and similar for human TG1 and mouse TG2 apart from enzyme concentration (mTG2 at 5 nM; TG1 at 10 nM) and CaCl2 concentration (0.2 mM for mTG2; 0.05 mM for TG1). Factor XIII was activated using 0.1 µg/µL thrombin (Sigma) in 35 mM Tris (pH 8.0) for 20 min at 30 °C, and the transamidation reaction was performed with 20 nM factor XIIIa in 50 mM Tris (pH 8.0), 1.25 mM CaCl2, 0.05% Pluronic F-127, and 0.2 mM DTT. TG3 was activated with 0.02 µg/µL thrombin under the same conditions as factor XIII, and assay conditions were 10 nM TG3 in 50 mM HEPES (pH 8), 20 mM CaCl2, 0.2 mM DTT, and 0.05% Pluronic F-127.
Radioactive transamidation assay
Transamidation activity in a nonfluorescent format was performed as described 23 with slight modifications. Incorporation of 300 nM [3H]-putrescine (GE Healthcare Life Sciences, Uppsala, Sweden) into NMC catalyzed by 20 nM TG2 was measured under conditions as used for the fluorescent assay described above in a total of 36 µL using a 96-well plate. After 60 min, the reaction was stopped by addition of 240 µL ice-cold 10% trichloroacetic acid (TCA) and incubated for 1 h at 4 °C. The solution was then applied to a filter plate and rinsed twice with 3% TCA and allowed to dry overnight at 37 °C. A total of 30 µL scintillation fluid was added per well, and radioactivity was quantified with a Top-Count™ (PerkinElmer, Waltham, MA).
Caspase-3 biochemical assay
The caspase-3 counterassay is based on cleavage of the caspase-3-specific peptidic substrate Z-DEVD-R110 (Invitrogen) at 20 µM. As a reference inhibitor, Ac-DEVD-cho was used. The reaction was performed using 0.05 U/µL caspase-3 (Biomol) in 50 mM HEPES (pH 7.4), 100 mM NaCl, 0.1% Chaps, 1 mM EDTA, 10% glycerol, and 0.2 mM DTT with a 30-min incubation time. An endpoint measurement was recorded using a microplate reader (Safire or Ultra, Tecan; ex: 485 nm, em: 535 nm).
Cellular assays
A HEK cell line with stable TG2 overexpression has been generated using a human TG2 construct kindly provided by Prof. G. V. Johnson. 24 Seventy-five thousand cells were seeded per well in a 96-well plate and allowed to attach overnight. The cell medium was then discarded, and serum-free medium containing 20 µM retinoic acid was added to increase TG2 activity. Test compounds were added with a final DMSO concentration of 0.5%. After 18 to 22 h, 0.33 µCi [3H]-putrescine was added per well and incubated for 2 h at 37 °C. For intracellular TG2 stimulation, final concentrations of 10 mM CaCl2 and 1 µM calcimycin were added and incubated for 1 h at 37 °C. After 2 wash steps, cells were lysed for 30 min at room temperature (RT) in 60 µL of 50 mM Tris (pH 7.5), 300 mM NaCl, 1 mM EDTA, and 0.5% Triton X-100. The lysate was precipitated by adding 100 µL TCA (10%), and the plate was mounted on an automatic harvester, washed (3.3% TCA), dried, and counted.
Cytotoxicity assay
Compounds were applied to the cells as described above, and on the assay day, ready-to-use ATPlite (PerkinElmer) solution was added, and the readout was developed as instructed by the supplier.
GTP binding assay
Binding of a fluorescent GTP probe was measured as described. 2 In short, 50 nM Bodipy-GTPγS (Invitrogen) and 0.5 µM TG2 with or without compounds were allowed to bind under assay conditions as described for the above TG2 transamidation fluorescent assay but in the absence of CaCl2. The samples were measured on a microplate reader (Safire or Ultra, Tecan; ex: 490 nm, em: 523 nm).
Compound and reagent handling
Compounds were dissolved in 100% DMSO at 10 mM and stored at 4 °C for frequent use or at −20 °C for long-time storage. For concentration responses, initial dilution series were prepared in 100% DMSO. For the biochemical assays, enzyme and compounds were preincubated for 15 min at 30 °C, and the reaction was started by adding the substrate. For inactivation kinetics of irreversible inhibitors, enzyme was added to a mixture of inhibitor and substrate, and a continuous measurement was started immediately.
Data analysis
IC50 values were determined with an automated in-house data evaluation software, A+, 25 using a hyperbolic binding model. All data points were normalized between 0% and 100% inhibition using the appropriate positive and negative controls. Kinetic data for inactivation measurements were analyzed using GraphPad Prism as described. 26 In short, the observed inactivation rate, kobs, was determined by an exponential fit of the raw data (RFU = relative fluorescent units) according to RFU = RFUmax · [1 − exp(−kobs · time)] + offset. The inactivation efficiency kinact/KI was determined from a linear fit of kobs versus the inhibitor concentration.
Results
TG2 inhibitor profiling platform
We have used our platform of screening assays to profile and characterize several reported TG2 inhibitors, including transamidation site inhibitors as well as GTP site inhibitors, and have defined their potency, efficacy, and selectivity. For comprehensive characterization, we established a platform of primary and secondary assays as well as selectivity assays that assess transglutaminase inhibition, specificity, cellular activity, and mechanism of action.
Primary TG2 transamidation assays
In the initial analysis, inhibitory potency against human TG2 was measured in a cell-free setting (
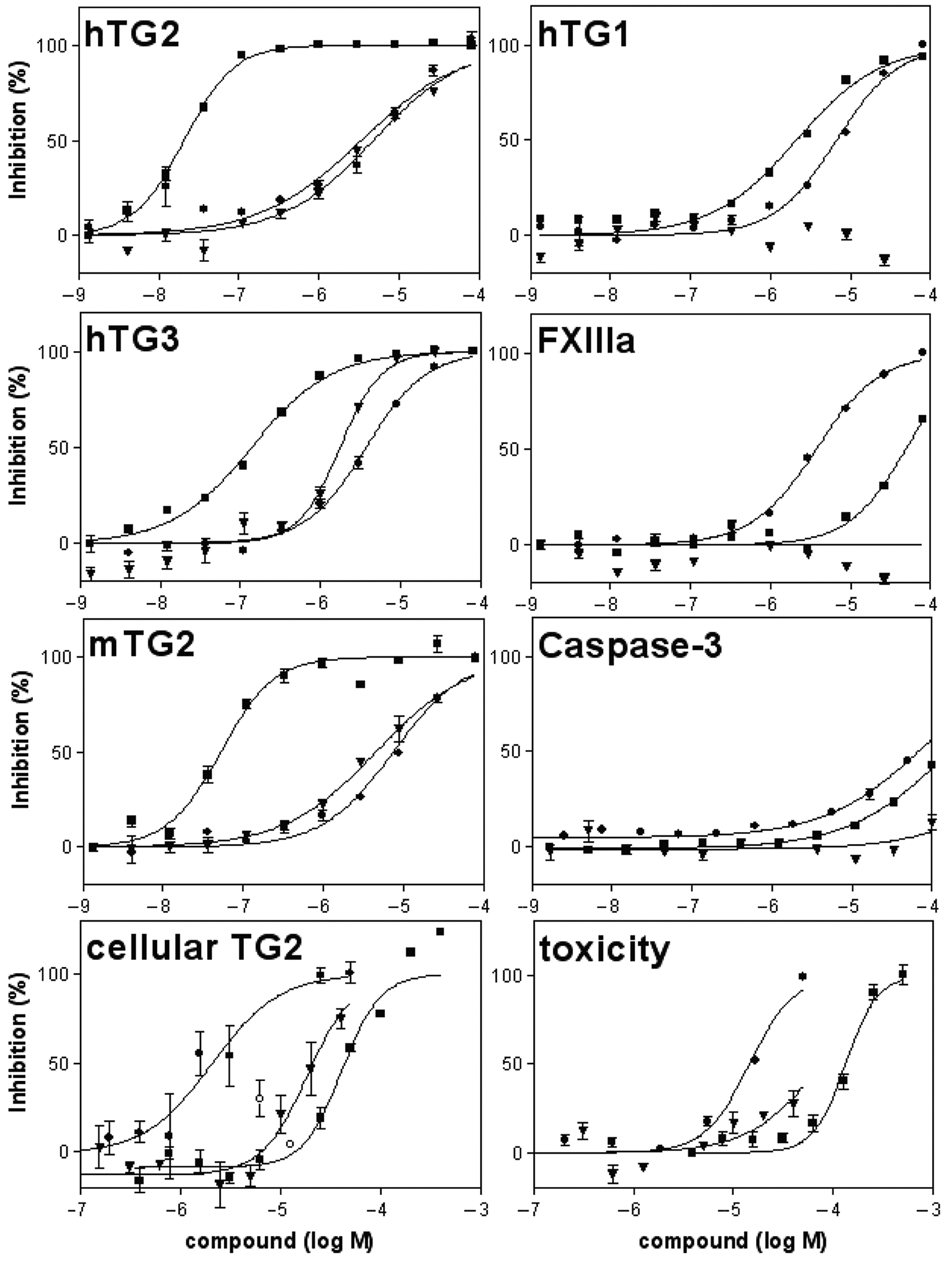
Exemplary concentration-response curves obtained in the platform assays: primary assay (hTG2), biochemical selectivity and counter- assays (hTG1, hTG3, FXIIIa, mTG2, caspase-3), and cellular assays (cellular TG2 inhibition and toxicity assay). Selected compounds are shown: irreversible peptide inhibitor Z-DON (■), hit from library screen GW 5074 18 (●), and CHDI-00209424 (novel quinazolinone) (▼). GW 5074 shows a characteristic and reproducible trace in the cellular assay, with 50% inhibition at 3 and 1.5 µM followed by a potency drop at 6 and 12 µM (○) and toxicity at 25 and 50 µM, indicating multiple effects in the intracellular environment.
Selectivity assays
For compounds with an IC50 < 5 µM against hTG2, selectivity against human TG1, TG3, factor XIIIa, and mouse TG2 was assessed (
Secondary TG2 transamidation cellular assays
Selective and potent compounds were tested for their inhibitory activity in a cellular TG2 transamidating assay using a novel assay based on incorporation of radioactive putrescine in HEK cells with stable TG2 expression (
Mechanism-of- action assays
To identify the binding mode of novel or selected compounds, various assay parameters were altered to address different binding properties.
Reversible compounds were investigated for binding to the active site by titrating one substrate while the other was fixed at its Km. Competitive binding properties are indicated by a Km increase, whereas the Vmax is not changed.
GTPase and calcium-activated transamidation are enzymatic properties of TG2 that are controlled by the conformation and exclude each other to a certain extent. At the calcium concentration with half-maximal transamidation activity, which has been determined for all isoforms and adjusted accordingly in the assays (
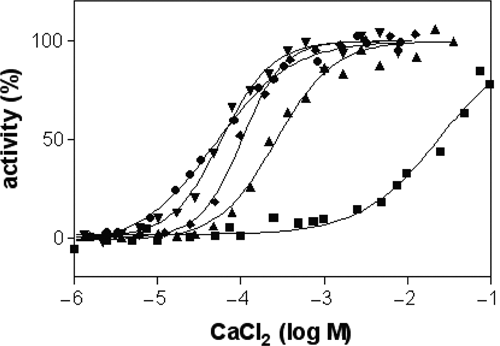
Calcium activation of different transglutaminase (TG) isoforms. Reaction velocities measured at each calcium concentration were normalized between 0% and 100% activity. Concentrations of CaCl2 with half-maximal activity (EC50) were 0.27 mM for human TG2 (▲), 0.06 mM for TG1 (▼), 25 mM for TG3 (■), 0.05 mM for FXIIIa (●), and 0.1 mM for mouse TG2 (♦).
The compounds were assayed for irreversible binding by introducing a preincubation step of enzyme and inhibitor 10-fold above its IC50, followed by a 100-fold dilution and direct measurement of enzyme activity. Irreversible binding is indicated if enzyme activity cannot be restored even though the compound has been diluted 10-fold below its IC50. It should be noted that compounds with a very slow dissociation reaction in the range of the assay time (~30 min) appear as irreversible inhibitors. For irreversible inhibitors, further experiments are required to determine the inactivation rate (kobs) of the enzyme caused by inhibitor binding. 29 By plotting kobs versus the inhibitor concentration, the second-order rate constant (kinact/KI) can be obtained, which is a suitable measure of inhibitor efficiency. If this parameter is influenced by varying substrate concentrations, binding to the active site is likely. For irreversible inhibitors, the binding site may also be identified using tryptic digestion followed by mass spectrometry.
Reversible transamidation site inhibitors/pseudo-inhibitor substrates
Many commonly used TG2 inhibitors such as putrescine, cadaverine, spermidine, and cystamine
30
are in fact alternative substrates to the tracer (i.e., labeled putrescine or KxD) used in cellular and/or biochemical TG2 transamidation assays. They should therefore be considered as pseudo-inhibitor substrates for the reaction. Cystamine has competitive properties toward KxD as indicated by a Km increase with unchanged Vmax (data not shown), which is expected for an alternative substrate. It also does not show a significant preference for TG2 compared to TG1, TG3, or FXIIIa (
IC50 Values Obtained in the Platform Assays
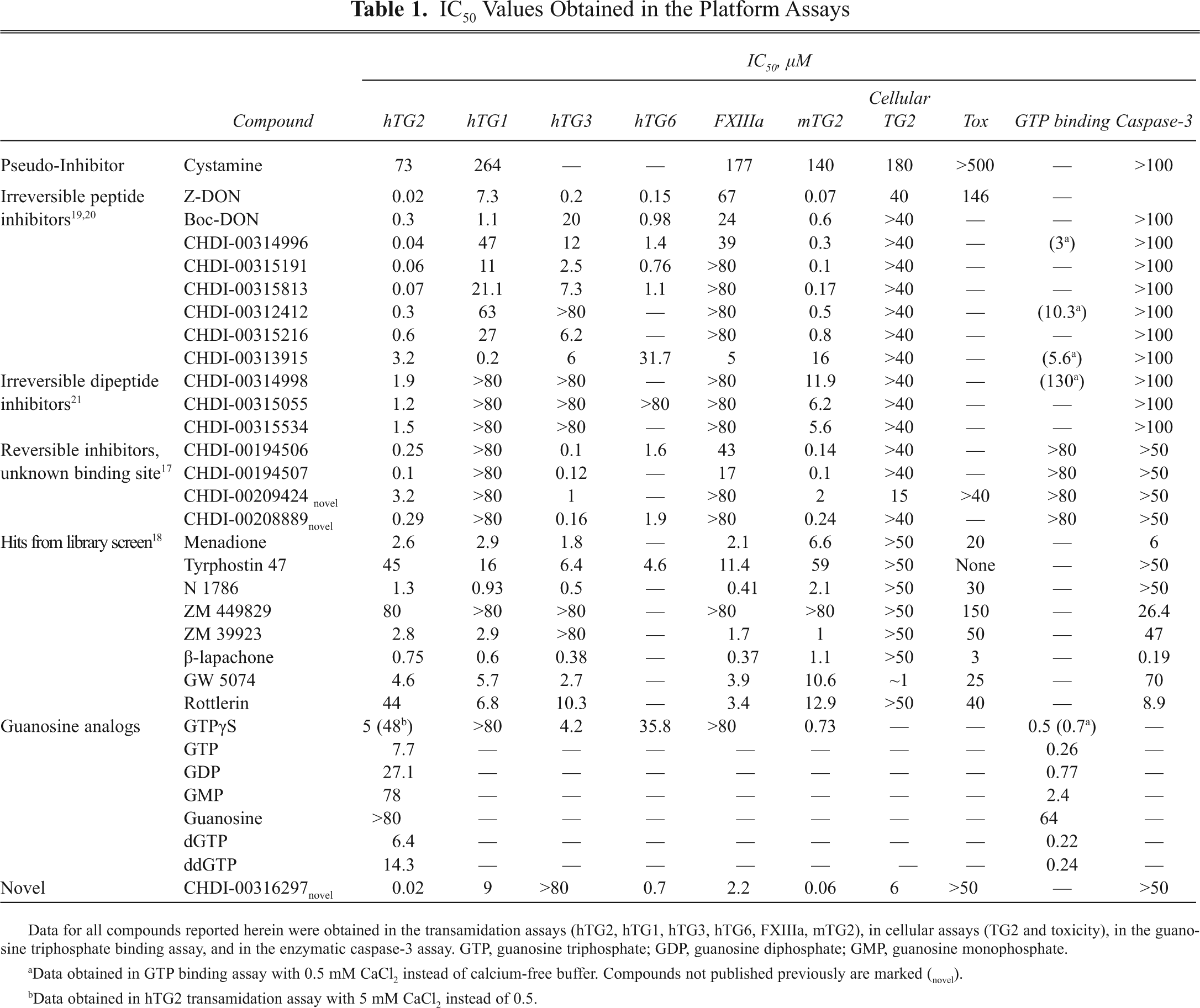
Data for all compounds reported herein were obtained in the transamidation assays (hTG2, hTG1, hTG3, hTG6, FXIIIa, mTG2), in cellular assays (TG2 and toxicity), in the guanosine triphosphate binding assay, and in the enzymatic caspase-3 assay. GTP, guanosine triphosphate; GDP, guanosine diphosphate; GMP, guanosine monophosphate.
Data obtained in GTP binding assay with 0.5 mM CaCl2 instead of calcium-free buffer. Compounds not published previously are marked (novel).
Data obtained in hTG2 transamidation assay with 5 mM CaCl2 instead of 0.5.
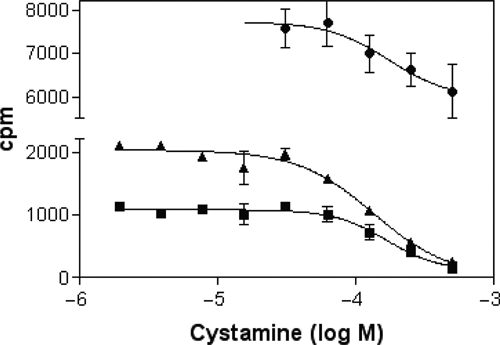
Incorporation of radioactively labelled amines into cellular protein and its inhibition by cystamine. Cellular TG2 assay signal under standard conditions with [3H]-putrescine, IC50 = 170 µM (■); [3H]-putrescine + 100 µM verapamil (applied together with cystamine), IC50 = 140 µM (▲); [3H]-spermidine, IC50 = 180 µM (●). In all cases, 0.3 µCi was applied per well.
Irreversible transamidation site inhibitors
Irreversible inhibitors display slow inactivation kinetics, being limited by formation of the covalent bond, and inactivation kinetics has to be measured to assess compound efficiency. However, the good correlation of IC50 values versus kinact/KI determined for the irreversible inhibitors used in this study indicates that the IC50 is a good measure of compound potency (
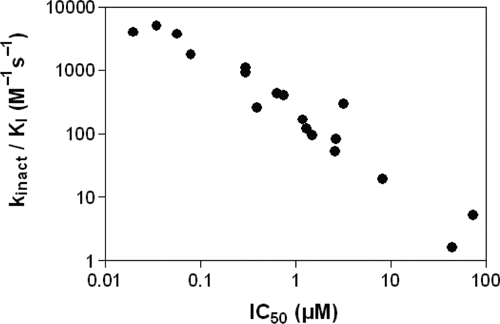
Correlation of kinact/KI values versus IC50. Inactivation efficiency kinact/KI was derived for all irreversible inhibitors reported herein from a linear fit of kobs versus inhibitor concentration and plotted against the IC50 values determined under standard conditions.
Various published and commercially available irreversible peptide inhibitors have been characterized for selectivity and cellular activity (
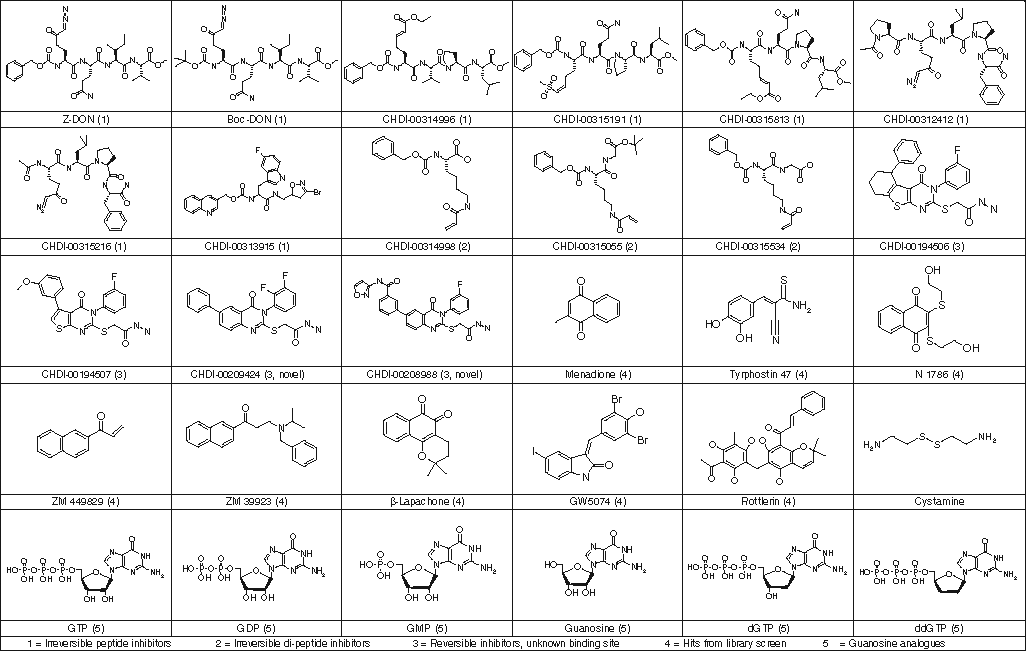
Structures of compounds described in Table 1.
A set of irreversible dipeptide TG2 inhibitors published by Marrano et al.
21
has been synthesized and tested in all available assays (
Hydrazide TG2 inhibitors
A set of TG2 inhibitors based on a quinazolinone hydrazide scaffold was identified by Duval et al. 17 An extensive characterization of these compounds has been published, indicating that they display slow binding kinetics to the GTP site itself or to an allosteric site that regulates GTP binding. 22 Analogs of these compounds have been synthesized, and slow binding kinetics and noncompetitive inhibition toward the substrates were confirmed for CHDI-00194506 (data not shown). However, these compounds did not compete with the binding of a GTP probe, indicating no direct binding at the GTP site. They showed good selectivity against TG1 and factor XIIIa but, in contrast, an even higher potency for TG3 than TG2. No cellular TG2 inhibition or toxicity was detected.
A large set of analogs was synthesized based on the hydrazide structure, and in addition novel quinazolinone compounds were prepared. Two examples of novel compounds, CHDI-00209424 and CHDI-00208889, are shown (
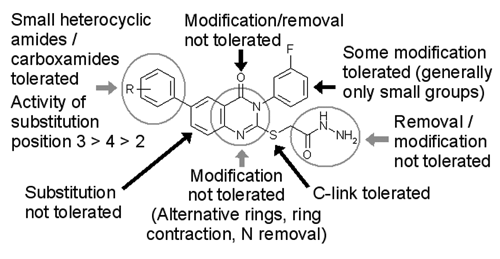
Medicinal chemistry evaluation of quinazolinone hydrazide inhibitors.
Compounds from pharmacologically active collection screens
Two libraries of pharmacologically active compounds (LOPAC and Prestwick) were screened by Lai et al.
18
for TG2 inhibition. (
Therefore, consistent with Lai et al., 18 we concluded that the compounds were unspecific, having general reactivity toward the active site cysteine residue.
GTP site inhibitors
Several guanine-based nucleotides were characterized for GTP site binding and transamidation inhibition (
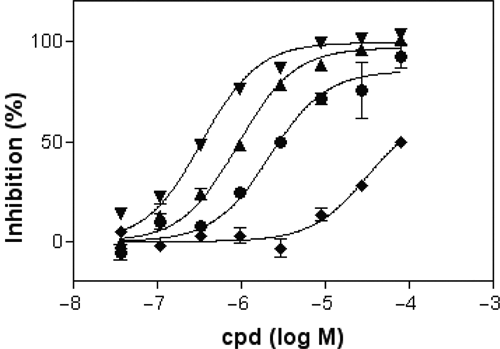
Inhibition of bodipy-GTPγS binding to TG2 (“GTP binding assay”). Concentration-response curves with guanosine triphosphate (GTP; ▼), guanosine diphosphate (GDP; ▲), guanosine monophosphate (GMP; ●), and guanosine (♦); IC50 values are shown in Table 1.
Competition with the GTP probe has been measured for a set of irreversible peptide inhibitors in the absence and presence of CaCl2. These inhibitors bind to the active site and should therefore not compete directly with GTP binding, but it has been shown with a crystal structure that CHDI-00312412 can lock TG2 in its open state
20
in the presence of CaCl2, which would lead to loss of GTP binding. In agreement with this model, the irreversible peptidic inhibitors CHDI-00314996, CHDI-00312412, and CHDI-00313915 (
Interestingly, in a
Novel TG2 inhibitors for testing in HD mouse models
Novel inhibitors are currently being developed that display sub-micromolar activity and good selectivity. Our initial hit compound for our lead series started by displaying greater than 100-fold selectivity over TG3 and approximately 10-fold selectivity over TG1 and TG6. It also showed moderate cellular activity and low toxicity. This series has been expanded and further developed to identify improved compounds with single-digit nanomolar IC50 for TG2 and at least 10-fold selectivity over all isoforms as well as low micromolar activity on cellular TG2 without toxic side effects (data not shown). Further development of this series has led to the discovery of a highly potent (20 nM) and selective inhibitor, CHDI-00316297 (
Discussion
In this study, we have profiled and further characterized TG2 inhibitors with different modes of action.
One example included reversible and allosteric inhibition by a hydrazide compound, but an extended medicinal chemistry program did not result in improvement of potency below 1 µM or of selectivity over TG3. Also, cellular potency was obtained only in rare cases. The binding site of these compounds could not be identified, and the work on this series was therefore suspended.
We have characterized various potent and irreversible peptidic inhibitors targeting the active site by competing directly with the glutamine substrate. Given that the initial contact between the active site cysteine residue and the first substrate involves a thioester bond formation, it is conceivable that potent inhibition can only be achieved by formation of an irreversible bond between cysteine and the inhibitor itself. The peptidic inhibitors that were tested showed good selectivity as would be expected for larger molecules, but the dipeptidic inhibitors also displayed very good selectivity over all TG isoforms tested herein. Concerns over the formation of drug-protein adducts aside, the examples shown in this study indicate that surprising selectivity profiles can be obtained with compounds as small as dipeptides. In addition, in a protein as large and complex as TG2, knowing the site of binding will greatly facilitate lead optimization efforts.
We have shown that cystamine is a reversible and unselective competitor of the lysine substrate, which is a profile expected for a pseudo-substrate inhibitor. Other examples of such competitors are putrescine, cadaverine, spermidine, and spermine. Thus, reversible, potent, and selective amine competitors that are not substrates for TG2 may be another field of interest for developing tools for in vivo studies.
Another approach toward TG2 inhibition is to populate the GTP site and thus to lock the enzyme in its closed and inactive state as described by Pinkas et al. 20 So far, GTP site binders other than guanosine-based nucleotides could not be identified so that selectivity issues with other GTPases could not be addressed. Also, the significant decrease of potency that follows the loss of phosphate groups when comparing the IC50 values generated for GTP, guanosine diphosphate (GDP), and guanosine monophosphate (GMP) indicates that this hydrophilic tail adds an important anchor. Compounds that mimic these properties may have a low chance of penetrating a cellular wall as well as the blood-brain barrier. However, inhibition of the GTP site is of great interest to evaluate simultaneous loss of transamidation and GTPase function.
Last, we characterized a set of unspecific inhibitors that are highly reactive toward thiol groups and therefore inactivate enzymes such as TG2 by binding to the active site cysteine. Such compounds are often obtained from screening of high-throughput screening compound libraries, but they can be easily identified by nonselective inhibition of other TG isoforms or other enzymes containing active site cysteine residues such as caspases. Given a robust means to identify such undesired compounds, screening of large compound collections remains a viable approach for the discovery of novel competitive or noncompetitive TG2 inhibitors.
In this study, we have presented a comprehensive set of assays for the characterization of TG2 inhibitor selectivity, cellular potency, and mechanistic properties. We have used this platform to generate profiles of several TG2 inhibitors that have been published or developed further in our medicinal chemistry program. We are currently optimizing a novel compound series that may provide us with a brain penetrant proof-of-concept inhibitor for testing in HD animal models to evaluate the series for potential for clinical application.
Footnotes
Acknowledgements
We thank Chris Yarnold and Fred Brookfield for valuable discussions, Jeanette Reinshagen, and Gerhard Schmiedel for technical support and Richard Marston, Jordan Palfrey, and Siw Johnsen for compound synthesis.