
Abstract
A cell-based fluorescent protein reporter assay for proteinase activity amenable to high-throughput applications was developed. This assay is based on Förster resonance energy transfer (FRET) between 2 variants of the green fluorescent protein connected by a short cleavable linker and expressed in Escherichia coli as tagged proteins. A library to assay proteinase specificity was generated by randomizing a portion of the linker using PCR. The library could be grown in microplates, allowing cells to be lysed in situ and substrate cleavage to be monitored through loss of FRET signal using a plate reader. Progress curves were generated to estimate cleavage efficiency, facilitating the identification of well-cleaved substrates. The polyhistidine-tagged fluorescent substrates could then be purified and used for further characterization. To establish the general utility of the screen, it was used to demonstrate that the cysteine proteinase of the hepatitis A virus, 3Cpro, prefers Ile, Val, or Leu at the P4 position of the cleavage sequence and Gly, Ser, or Ala at the P′1 position. The assay can also be used to screen small-molecule libraries for inhibitors.
Introduction
P
A variety of biological screens have been developed using fluorescent proteins. For example, caspase activation was studied by monitoring the cleavage of a concatenated pair of green fluorescent protein (GFP) variants where cleavage resulted in a loss of Förster resonance energy transfer (FRET) between the 2 variants. 7 A second example, CLiPS, 8 involved mixing a library of bacterial cells expressing peptide substrates on the surface with purified proteinase and a fluorescent tag, which recognized the N-terminus of the peptide. Screening comprised several rounds of (1) prescreening to remove cells not expressing substrate, (2) cleavage by incubating cells with proteinase, and (3) enriching for processed substrates using fluorescence-activated cell sorting (FACS).
The 3Cpro is a cysteine proteinase present in all Picornavirales viruses. 9 These viruses cause a variety of diseases in plants and animals that can seriously affect human health and agriculture. The 3Cpro is essential for the survival of the virus, processing the polyprotein. Understanding the specificity of 3Cpro would help identify proteinase cleavage sites, clarify the proteinase’s role in the viral life cycle, and facilitate development of proteinase inhibitors.
Herein, we present a high-throughput method for investigating proteinase specificity based on change in FRET signals of cells expressing a protein substrate consisting of 2 GFP variants. The method was validated by investigating the specificity of the hepatitis A virus (HAV) 3Cpro.
Materials and Methods
Reagents
Oligonucleotides were from Integrated DNA Technologies (Coralville, IA). Restriction enzymes, Antarctic phosphatase, and Taq DNA ligase were from New England Biolabs (Ipswich, MA). T4 DNA ligase was from Fermentas (Glen Burnie, MD). PCR was performed using the expand High Fidelity DNA polymerase system. All other chemicals were of analytical grade. Nucleotide sequencing was performed at the NAPS unit of the University of British Columbia. DNA was propagated and cloned using Escherichia coli DH5α (see
Molecular cloning
The ypet and cypet genes were cloned into a derivative of pBAD24 in which the BamH1 restriction site outside of the multiple cloning site had been changed to a unique NotI site. The latter mutagenesis was performed using oBAD(Bam-Not) (see
The cypet gene was amplified by PCR from pCyPet-His, 10 using primers oCyFor and oCyRev. The ~0.8-kb amplicon was digested with KpnI and EcoR1 and cloned into pBAD24X to yield pBXCy-H, which encoded a polyhistidine-tagged (ht-)CyPet. The ypet gene was similarly amplified from pYPet-His using oYFor and oYRev, digested with PstI and EcoR1, and cloned into pBAD24X to yield pBXYP-H, which encoded ht-YPet.
The ypet gene was excised from pBXYP-H with enzymes KpnI and PstI and inserted into pBXCy-H to yield pBXCyY-H. This plasmid carries genes encoding ht-CyPet and a nontagged YPet on a single transcript under the control of the PBAD promoter. A translation-enhancing region and a Shine-Dalgarno sequence between the genes ensure translation of ypet.
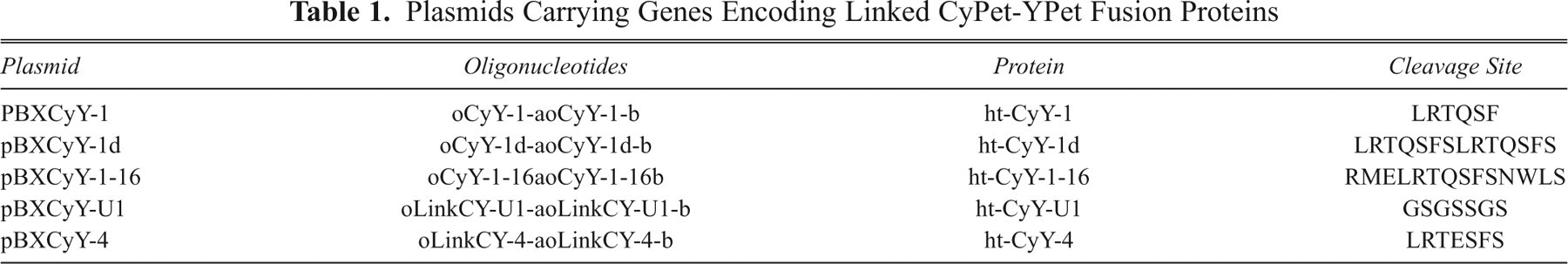
Fluorescent fusion proteins were generated by inserting a short linker consisting of 2 synthetic semi-complementary oligonucleotides into plasmid pBXCyY-H. The linkers were generated by heating and slowly cooling an equimolar mixture of the oligonucleotides to yield duplexes with 3′ and 5′ overhangs. Plasmid pBXCyY-H was digested with BamH1 and KpnI and ligated with duplex-forming oligonucleotides oCyY-1-a and oCyY-1-b. This yielded plasmid pBXCyY-1, which encoded ht-CyY-1. Other constructs were similarly generated using oligonucleotides listed in Table 1 .
Plasmids Carrying Genes Encoding Linked CyPet-YPet Fusion Proteins
To generate the libraries of plasmids encoding ht-CyPet and YPet connected by potentially cleavable linkers, the yfp gene was amplified from pBDXYFP using oYFP-revhis2 and either oLRTQXSF-YPet or oXRTQSFS-YPet. The sequence NN(G/T) was used to encode a random amino acid (X) at either the P4 or P′1 position of the cleavage sequence. The resulting ~0.8-kb amplicons were digested using BamHI and HindIII and inserted into pBXCy-H to yield libraries P′1 and P4, respectively.
Production and purification of fluorescent protein substrates and proteinase
HAV 3Cpro was produced and purified essentially as described. 11 For fluorescent protein production, a colony of freshly transformed E. coli BW27783 containing an appropriate plasmid was grown overnight in 50 mL LB ampicillin at 37 °C. Then, 1 L of LB was inoculated with 10 mL of overnight culture and grown at 37 °C. Fluorescent protein production was induced by adding arabinose to a final concentration of 0.2% when the culture attained OD600 of ~0.5. The cells were incubated overnight at 25 °C, harvested by centrifugation, and frozen at −80 °C.
To purify fluorescent proteins, cell pellets were resuspended in ~35 mL 20 mM sodium phosphate (pH 7.4), 500 mM NaCl, and 20 mM imidazole and lysed using a French press. Cell debris was removed by ultracentrifugation, and the supernatant was passed through a His GraviTrap (GE Healthcare, Piscataway, NJ). Eluted protein was exchanged into 100 mM potassium phosphate, 2 mM EDTA (pH 7.5) by ultrafiltration; concentrated to 5 to 30 mg/mL; frozen as beads in liquid nitrogen; and stored at −80 °C.
Fluorescence-based cleavage assays
Substrate cleavage was monitored fluorometrically in 100 µL 100 mM potassium phosphate, 2 mM EDTA (pH 7.5) at 37 °C using either a Varian Eclipse spectrofluorometer (Cary; Varian, Palo Alto, CA) and a quartz cuvette or a Victor 2 plate reader (PerkinElmer, Waltham, MA) and a 96-well black plate. Using the fluorometer, fluorescence was measured using λex = 434 nm and λem = 477 nm (for CyPet) or λem = 527 nm (for FRET), and initial rates were calculated using the first 5 min of the progress curve. Using the plate reader, fluorescence was monitored using settings for CyPet (λex = 430 nm, λem = 470 nm) and FRET (λex = 430 nm, λem = 535 nm), and initial rates were calculated from the first 45 min of the progress curve.
Screening of fusion protein library
E. coli BW27783 cells transformed with the appropriate plasmids were grown overnight at 37 °C on LB agar media with ampicillin and tetracycline. For each library, 96-deep well plates containing 1.5 mL of LB with ampicillin, tetracycline, and 0.2% arabinose per well were inoculated with a single colony. Cultures were grown overnight at 37 °C in a sealed Ziplock freezer bag filled with 100% O2, harvested by centrifugation, and stored at −80 °C. Lysates were prepared by resuspending thawed cells in 50 µL of Bugbuster™ (Novagen, Gibbstown, NJ) and incubating at room temperature for 20 min. After incubation, 350 µL of 100 mM potassium phosphate, 2 mM EDTA (pH 7.5) was added; the sample was centrifuged; and the supernatant was screened.
To measure cleavage, 100 µL of the supernatant was added to a black 96-well plate and prewarmed at 37 °C. Initial fluorescence of YPet, CyPet, and FRET was measured. The reaction was initiated with 3Cpro to a final concentration of 1 µM. Changes in CyPet and FRET signals were measured over 2.5 h.
Results and Discussion
Dependence of fusion protein cleavage on linker length
To optimize substrate cleavage for the screen, linkers of 3 different lengths and composition were compared: substrate ht-CyY-1 had linker sequence LRTQ/SFS derived from the 2A/2B cut site of the HAV polyprotein, substrate ht-CyY-1-16 had the extended sequence RMELRTQ/SFSNWLS, and substrate ht-CyY-1d had sequence LRTQ/SFSLRTQ/SFS. Each substrate was purified (>80% as measured by sodium dodecyl sulfate polyacrylamide gel electrophoresis [SDS-PAGE]), and the relative rate of cleavage by HAV 3Cpro (purified to >90% by SDS-PAGE) was determined. Substrate cleavage was confirmed by native-PAGE analysis. At 25 µM substrate, the relative cleavage efficiency of each was ht-CyY-1 (1.0), ht-CyY-1d, (1.6) and ht-CyY-1-16 (1.4), suggesting that linker length did not significantly affect rate of cleavage.
Detecting cleavage of fusion substrate in cell lysates
To test cleavage of fusion protein by exogenously added proteinase in raw extracts and cleared lysates, cells producing either ht-CyY-1 or an uncleavable variant, ht-CyY-U1, were grown in 96-well plates in O2-filled bags. Extra O2 ensured sufficient biomass and fluorescent protein production for the screen. Addition of 3Cpro to either raw extracts or cleared lysates resulted in a steady loss of FRET signal for cleavable compared to uncleavable substrates. Progress curves were similar for ht-CyY-1 in raw extracts and cleared lysates (data not shown).
The ability to use cell lysates rather than purified substrates presents several advantages. First, the library is easily generated using molecular cloning techniques, and by eliminating purification, larger libraries can be screened. Second, substrate cleavage can be measured continuously, facilitating identification of well-cleaved substrates using progress curves. Finally, normalized initial rates (loss of F527) can be used to approximate relative kcat/Km, as reported for CLiPS. 8 Nevertheless, this value is approximate as the screening is not conducted under steady-state kinetic conditions.
Optimizing of 3Cpro concentration for the screen
To optimize the concentration of proteinase used in the screen, up to 1 µM HAV 3Cpro was added to cleared lysates containing either ht-CyY-1 or ht-CyY-U1. Initial rates of fluorescence loss at 527 nm were plotted against the initial F527 signal (approximating fusion protein concentration) ( Fig. 1 ). For ht-CyY-1, the initial rate of cleavage, as measured by loss of F527, depended on both substrate and proteinase concentrations. At a fixed concentration of 3Cpro, rates increased nonlinearly with substrate concentration ( Fig. 1 , x-axis). Rates also increased linearly with proteinase concentration. Above 1 µM 3Cpro, there was significantly more error in the measured rates (results not shown). Some loss of F527 was observed for ht-CyY-1 in the absence of proteinase and for ht-CyY-U1 (data not shown). Interestingly, this loss was proportional to initial FRET signal and was similar to losses observed in individually expressed ht-CyPet or ht-YPet proteins (data not shown), suggesting a background loss of signal due to bleaching rather than proteolytic cleavage by an endogenous E. coli proteinase. Based on these results, 1 µM 3Cpro was used in subsequent screens as this concentration yielded significant differences in the rate of F527 decline due to cleavage over background.
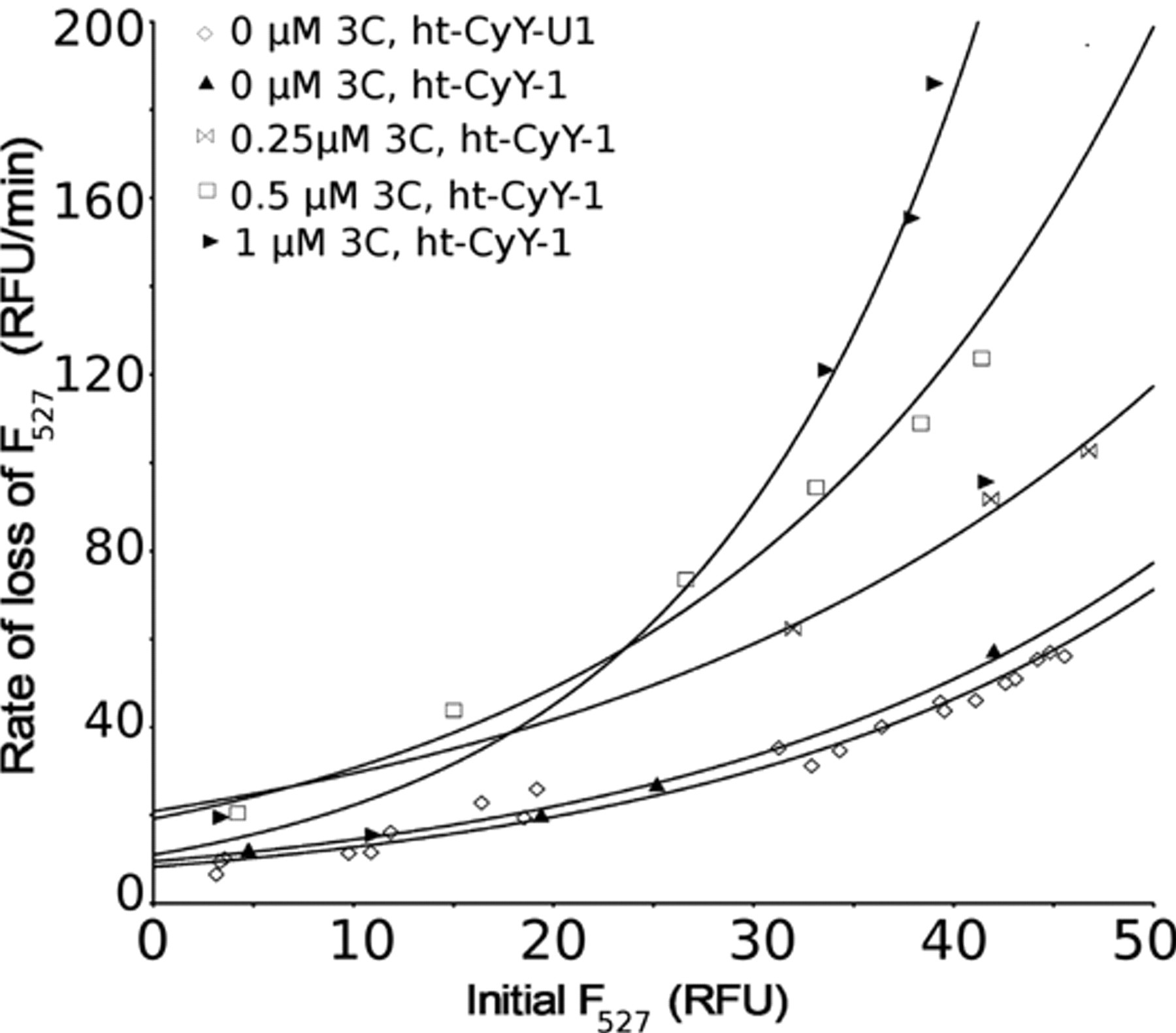
The dependence of cleavage rates of ht-CyY-1 on HAV 3Cpro and substrate concentrations. Initial Förster resonance energy transfer (FRET) signal is proportional to fluorescent protein concentration. Exponential curves were used to highlight trends in the data.
Screening the substrate preference of HAV 3Cpro
To validate the screen, the specificity of 3Cpro was screened using libraries of substrates in which either the residue at the P4 position of the cleavage sequence (XRTQ/SFS) or the P′1 position (LRTQ/XFS) was varied. Random sequencing of 100 clones confirmed libraries’ completeness (results not shown). Based on calculations, the screening of 145 clones provides a 99% chance that the least represented sequence is screened. 12
Library clones were grown in 96-well plates, and initial velocities were calculated using proteinase and cleared lysates as described above. Progress curves displaying obvious irregularities such as jumps in the baseline were eliminated. Initial velocities were plotted versus initial YPet signal instead of the initial FRET signal for 2 reasons. First, the YPet signal is likely to be less influenced by changes in linker structure between variants in the library. Second, measuring YPet signal directly enabled elimination of clones lacking YPet because of the presence of a stop codon in the linker.
In a plot of rate of loss of FRET signal versus YPet signal ( Fig. 2 ), cells containing CyPet (ht-Cy-H) lay along the y-axis as they had no significant F527 signal. Cells containing ht-CyY-U1 lay close to the x-axis as ht-CyY-U1 was uncleavable. Cells containing ht-CyY-1, a well-cleaved substrate, were positioned along a diagonal between the limits defined by ht-Cy-H and ht-CyY-U1. Library substrates were cleaved at rates between the cleaved and uncleaved controls, with none being faster than the wild-type.
These initial rates of cleavage were normalized to fusion protein concentration by taking the ratio of rate/YPet: clones with higher normalized rates presumably contain better cleavage sequences. Based on the normalized rate and clone position in the plot ( Fig. 2 ), 14 clones from the P4 library and 8 clones from the P′1 library were selected for sequencing to identify the randomized residue ( Fig. 3 ).
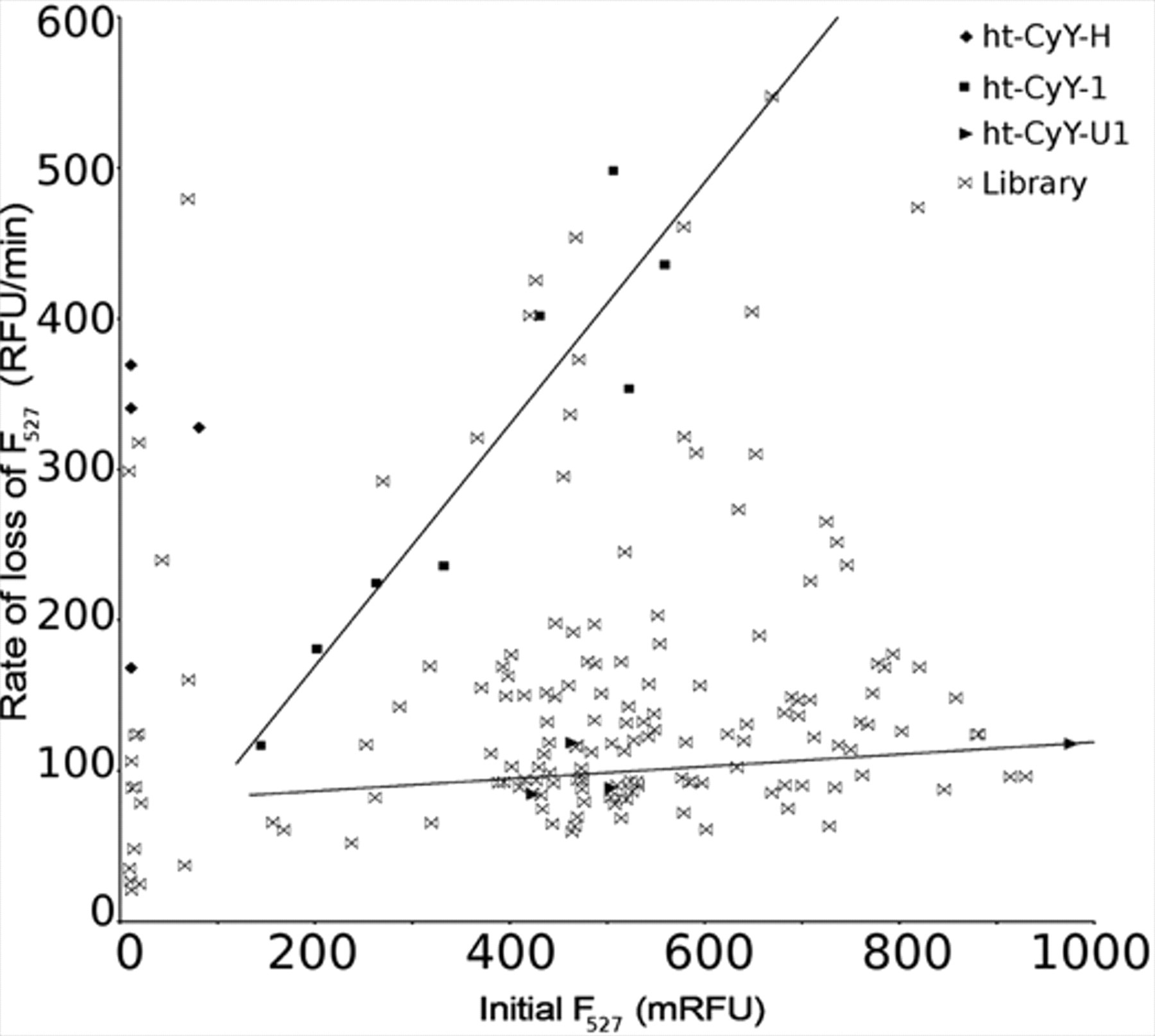
Screening of the P4 library for cleavage by HAV 3Cpro. Initial velocity is plotted against initial YPet signal. Trends for the ht-CyY-1 and ht-CyY-U1 are indicated by best-fit lines.
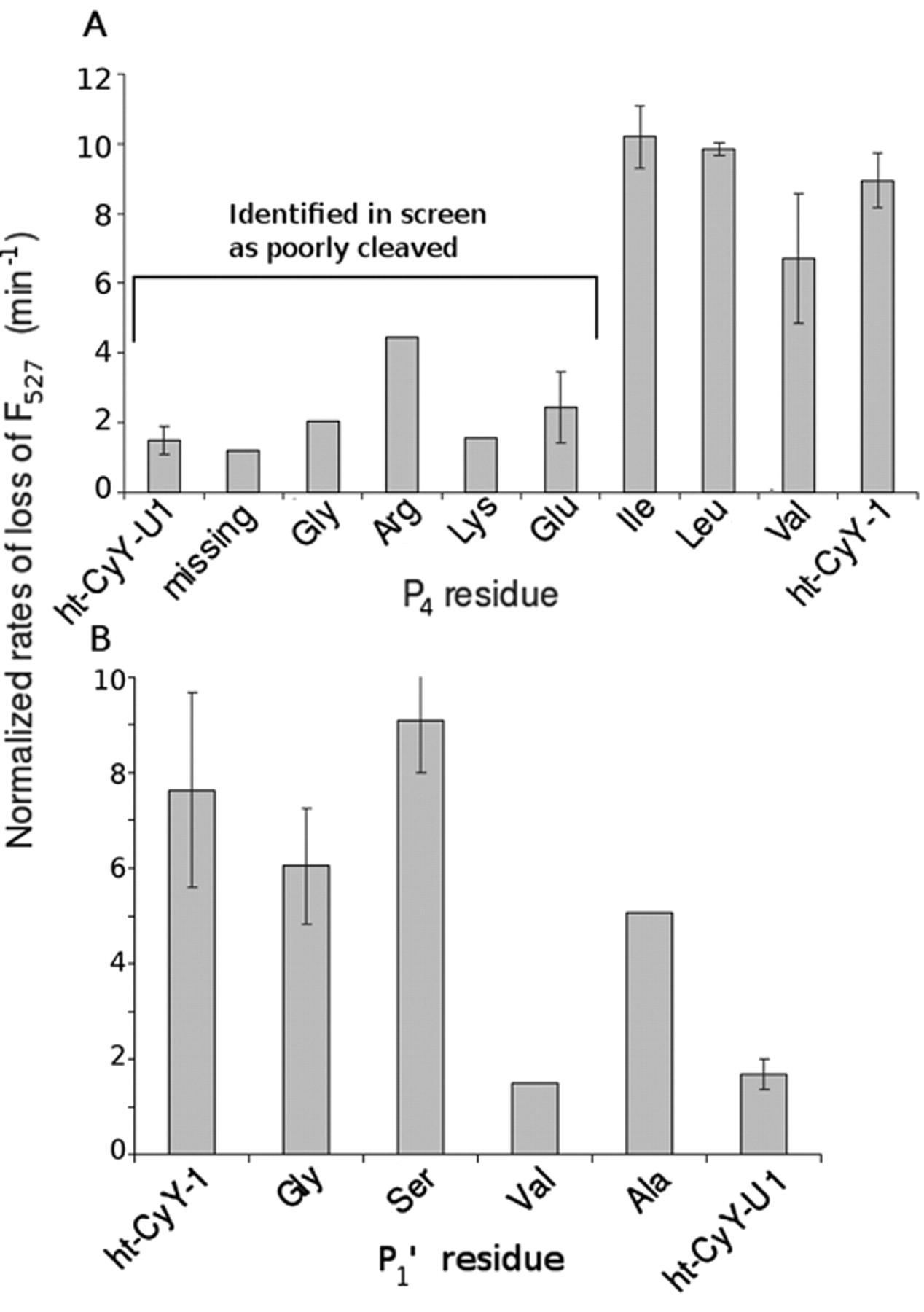
Rate of change in Förster resonance energy transfer (FRET)/YPet signals for selected clones from the HAV 3Cpro screen of cleared lysates of the (
The substrate preference of HAV 3Cpro determined using the screen is consistent with previous studies using peptides and with structural analysis. Specifically, the best substrates had Ile, Leu, or Val at the P4 position and Gly, Ser, or Ala at the P′1 position (
Fig. 3
). Substrates having either charged (Arg or Lys) or small (Gly) residues in the P4 position were poorly cleaved. In a study of 11 synthetic peptides with different natural and artificial amino acids in the P4 position, substrates with Leu, Trp, Val, Ile, and
Kinetic characterization of cleavage of P′1 position substrates
Three P′1 variants (ht-CyY-1A, -1G, and -1V, differing from ht-CyY-1 in having an Ala, Gly, and Val, respectively, at the P′1 position) were purified and kinetically characterized. At a concentration of 4.5 µM, substrates ht-CyY-1A and ht-CyY-1G were well cleaved relative to ht-CyY-1 (relative rates were 0.31 and 0.28, respectively; less than 5% error). The substrate with Val in the P′1 position was cleaved at a relative rate of 0.03, consistent with its identification in the screen as a poor substrate.
Conclusions
This article reports a novel high-throughput screen for analyzing proteinase activity using fused GFP variants. In contrast to peptide screens, libraries are readily generated by PCR amplification of ypet using an oligonucleotide encoding the linker library. Nevertheless, the fluorescent protein substrate may limit accessibility to the active site. The fluorescent protein screen is also rapid: unlike the CLiPS method, 8 the plate assay involves a single growth and cleavage step, enabling analysis to be completed within 24 h of library generation. Purification and validation of substrates identified from the screen are straightforward as substrates are polyhistidine tagged. Finally, the plate method can be expanded to screen libraries of proteinases by adding purified substrate to cell lysates or to screen small molecules for proteinase inhibitors.
Four challenges limit the widespread application of the described screen in its current form: (1) potential exosite influence, (2) potential confounding effects from endogenous E. coli proteinases, (3) the availability of the proteinase, and (4) liquid handling. Although the current method does not allow for the screening of exosites, similar to the other screens described herein, it may be possible to introduce such sites between the 2 GFPs. Although endogenous proteinases did not confound the current screen, the (non)cleavage of substrates is easily tested by comparing progress curves with and without exogenously added proteinase. With respect to the third limitation, this screen is potentially limited to proteinases that can be heterologously produced. Purification of the proteinase could be avoided by using lysate of cells, provided they contained sufficient proteinase activity. Indeed, similar proteinase specificity results were obtained using extracts of cells containing HAV 3Cpro (data not shown). Similarly, liquid handling could be reduced by eliminating the additional lysate-clearing step or by growing and lysing cells in the plate used for fluorescence measurements. Nevertheless, substrate production must be sufficient to distinguish cleaved from uncleaved substrates.
Footnotes
Acknowledgements
The ypet and cypet genes were a gift from Prof. Patrick S. Daugherty.
This work was supported by Natural Sciences and Engineering Research Council (NSERC) of Canada Strategic and Discovery grants to LDE. CH was the recipient of an NSERC PGS-D scholarship.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.