
Abstract
Objective:
To evaluate the real-world efficacy, safety, and functional outcomes of PRC-063 (multilayer-release methylphenidate) versus lisdexamfetamine (LDX) in ADHD subjects in a phase IV, open-label study.
Method:
The primary endpoint was the change in the ADHD-DSM-5 Rating Scale (ADHD-5-RS) total score from baseline to Month 4. Secondary endpoints included a non-inferiority comparison between PRC-063 and LDX and measures of functioning and evening behavior.
Results:
One hundred forty-three pediatric and 112 adult subjects were enrolled. Mean ADHD-5-RS scores (standard deviation) were reduced in pediatric (−16.6 [10.4]) and adult (−14.8 [10.6]) subjects treated with PRC-063 (p < .001). PRC-063 was non-inferior to LDX in the pediatric population but not in the adult population. Significant improvements were demonstrated in quality of life and functionality. Both medications were well-tolerated; more adverse events led to study discontinuation in pediatric subjects treated with LDX versus PRC-063.
Conclusion:
PRC-063 and LDX significantly improved ADHD symptomatology and functioning and were well-tolerated.
Introduction
ADHD is a common neurodevelopmental disorder, characterized by a persistent pattern of inattention, hyperactivity, and impulsivity, that is often first diagnosed in childhood and may persist into adulthood (Adamou et al., 2013; Wolraich et al., 2011). Impaired functioning due to ADHD has a wide-ranging impact on educational, interpersonal, physical, emotional, social behavior, school and/or work-related activities in children (Caci et al., 2014) and adults (Bernardi et al., 2012; Pitts et al., 2015). ADHD-related impairments can occur during waking hours and significantly impact activities that take place before, during, and after school and/or work (Faraone et al., 2019; Fuermaier et al., 2021; Sallee, 2015).
The clinical focus for treatment of ADHD has shifted from management of symptoms to improving functional impairments and outcomes, with treatment goals focusing on improving overall quality of life (QoL) (Antshel, 2015; Canadian ADHD Resource Alliance [CADDRA], 2020). Long-acting psychostimulants (methylphenidate [MPH] and amphetamine [AMP]) are recommended as first-line pharmacological agents for the treatment of ADHD in Canada (CADDRA, 2020). PRC-063 (Foquest®) is a long-acting, once-daily, extended-release formulation of MPH with a rapid onset of action at 1-hr post-dose (A. C. Childress et al., 2020; A. Childress et al., 2022) and an extended duration of efficacy up to and including 16 hr post-dose in adults (A. Childress et al., 2022) and 13 hr post-dose in children (A. C. Childress et al., 2020). Prior to PRC-063, the duration of action for a long-acting stimulant had been demonstrated up to 14 hr post-dose in adults by lisdexamfetamine dimesylate (LDX, Vyvanse®) (T. Wigal et al., 2010). LDX is an AMP prodrug with an onset of action within 2 hr (1.5–2 hr) and a duration of action up to and including 13 hr post-dose in children (A. C. Childress et al., 2020) and 14 hr post-dose in adults (T. Wigal et al., 2010).
Although randomized controlled trials (RCTs) have demonstrated the efficacy, safety, and functional outcomes for both PRC-063 (A. C. Childress et al., 2020; A. Childress et al., 2022; Weiss, Cutler et al., 2021; Weiss, Surman et al., 2021; S. B. Wigal et al., 2020) and LDX (Adler et al., 2008; T. Wigal et al., 2010) compared to placebo, there is a paucity of head-to-head trials, and no published comparative real-world studies. While RCTs are considered the “gold-standard” of evidence generation, the applicability of their data to standard clinical practice can be limited by the closely controlled patient population, which often eliminates common comorbidities and other confounders. However, real-world evidence (RWE) studies enroll patients from standard clinical practice to provide information on comparative effectiveness, tolerability of therapies, pharmacoeconomic, and patient-reported outcomes and may provide insight with additional therapeutic benefits or uses beyond those studied in RCTs (Baumfeld Andre et al., 2020). Unlike typical RCTs, the patient population of a RWE study may include a more inclusive patient population which is more relevant to that seen in clinical practice.
The objective of this RWE, open-label study was to measure the change in ADHD symptoms with PRC-063 over a 4-month period across the lifespan of ADHD, including both pediatric (6–17 years old) and adult (≥18 years of age) subjects. A key secondary objective was a non-inferiority comparison of ADHD symptom improvements due to treatment with PRC-063 compared to that of LDX. Other secondary objectives examined how resultant symptoms affect different domains of functioning, including morning and evening behavior, as well as QoL.
Method
Study Design
This was a 4-month, phase IV, open-label, non-randomized study conducted at 13 Canadian sites. The first pediatric and adult subjects were enrolled in September 2019, and the last subjects completed in July 2021. Eligible subjects were assigned to a treatment group to receive either PRC-063 (25, 35, 45, 55, and 70 mg/day for all subjects and 85 and 100 mg/day for adult subjects only) or LDX (10, 20, 30, 40, 50, and 60 mg/day) and titrated to optimal dosage based solely on the investigator’s clinical judgment. At monthly visits, subjects were evaluated for their ADHD symptomatology by the Investigator (ADHD-Rating Scale-DSM 5 Version [ADHD-5-RS]) and completed questionnaires on functional outcomes (Weiss Functional Impairment Ratings Scale–Parent [WFIRS-P] or Weiss Functional Impairment Ratings Scale–Self [WFIRS-S]), morning and evening behaviors (Daily Parent Rating of Evening and Morning Behaviors–Revised [DPREMB-R] or Adult ADHD QoL Rating Scale–Revised [AAQoL-R]) and Patient Sleep & Satisfaction survey (PSS). Safety was evaluated through non-directed spontaneous adverse event (AE) reporting. Subjects were monitored for signs of suicide-related behavior, as per standard of care. A post-study safety follow-up phone call was conducted between 7 and 14 days after their last visit to assess subjects for any AEs that occurred following their last dose of medication received as part of the study. Following the last dose of study medication, the subject’s continuing, post-study ADHD treatment was based on investigator discretion and standard of care.
All documentation and procedures related to the study were executed in accordance with Good Clinical Practice (GCP) E6(R2) guidelines as required by the Declaration of Helsinki and guidelines for the “Conduct of Clinical Investigations” issued by the Therapeutics Products Directorate Canada (1997) and the Tri-Council Policy statement on Ethical Conduct for Research involving Humans (TCPS2-2014).
Subjects
Enrollment was open to male or non-pregnant, non-nursing female subjects aged 6 or greater with a diagnosis of ADHD (inattentive, hyperactive/impulsive, or combined type) who were deemed eligible to receive treatment with PRC-063 or LDX per their respective Canadian Product Monographs. Subjects aged 6 to less than 18 were eligible for the pediatric arm and those aged ≥18 years of age were eligible for the adult arm. To be eligible for the study, subjects, or their guardians if subjects were under 18 years of age, were required to be mentally and physically competent to provide informed consent or assent, and able and willing to comply with the study protocol, including the study duration.
Potential subjects who met any of the contraindications or warnings detailed in the respective Canadian Product Monographs were excluded from participation in the study. Other key exclusion criteria included: true allergy to MPH or AMPs or sympathomimetic amines, history of serious adverse reactions to MPH or AMPs, or previously non-responsive to MPH or AMP treatment (non-response was defined as MPH and AMP use at various doses for a phase of ≥4 weeks at each dose with little or no clinical benefit in the past 10 years); females of child-bearing potential who were pregnant, planning on becoming pregnant or breast feeding; subjects with a history of hyperthyroidism, thyrotoxicosis, advanced arteriosclerosis, seizures, severe renal insufficiency or glaucoma; subjects with structural cardiac abnormalities, symptomatic cardiovascular disease or moderate to severe hypertension; subjects who were receiving or within the past 14 days, received MAO inhibitors; a primary diagnosis of bipolar disorder, as assessed at baseline; investigational drug use (current or in the previous month); history of drug or alcohol abuse or dependence; and subjects who were deemed a suicide risk by the investigator.
Study Assessments
Primary Endpoint
The primary endpoint was the mean improvement in ADHD symptoms based on the ADHD-5-RS Total Score at Month 4 as compared to baseline for subjects treated with PRC-063. The ADHD-5-RS is a clinician-rated scale that reflects current symptoms of ADHD based on DSM-5 criteria. It is a global assessment scale that measures the severity of symptoms from visit to visit, but is not utilized to assess symptoms of ADHD over the course of the same day. The ADHD-5-RS consists of 18 items that are grouped into two subscales (hyperactivity/impulsivity and inattention) (DuPaul et al., 2016).
Secondary Endpoints
The key secondary endpoint was a non-inferiority comparison of change from baseline to Month 4 in ADHD-5-RS Total score between subjects who received PRC-063 and those subjects who received LDX, analyzed separately for pediatric and adult subjects, and analyzed for the efficacy and per-protocol (PP) populations. Other secondary efficacy measures included: WFIRS-S, WFIRS-P, DPREMB-R, AAQOL-R, and PSS.
The WFIRS-S/P are measures of ADHD-related functional impairment and were collected at each visit (Visit 1 [baseline], Month 1, Month 2, Month 3, Month 4). The secondary endpoints of WFIRS-S/-P change from baseline to Month 4 in WFIRS-S/-P were evaluated for PRC-063 subgroups and change from baseline to Month 4 in WFIRS-S/-P was evaluated for PRC-063 versus LDX. The WFIRS-S is a self-evaluation of functional impairments. The use of the WFIRS-S before and after treatment can allow the clinician to obtain an assessment of the subject’s specific areas of difficulty. Sixty-nine questions assess the severity of impact of the subject’s ADHD behavior on clinically relevant domains of functioning. The WFIRS-S has been psychometrically validated in an ADHD population (Canu et al., 2020). The WFIRS-P is a parent-completed evaluation of functional impairments, measured over the past month. Fifty questions assess the severity of impact of the subject’s ADHD behavior on clinically relevant domains of functioning. The WFIRS-P has been psychometrically validated in an ADHD population (Gajria et al., 2015; Weiss, 2022; Weiss et al., 2018). Functional improvement is commonly defined as the percentage of patients who achieve the minimal clinically important difference (MCID) while functional remission is defined as the percentage of patients who normalize at treatment endpoint.
DPREMB-R and AAQoL-R were completed at each visit (Visit 1 [baseline], Month 1, Month 2, Month 3, Month 4). The secondary endpoints of change from baseline to Month 4 in DPREMB-R (for subjects aged 6 to <18) or AAQoL-R (for subjects aged 18 or older) were evaluated for PRC-063 subgroups and change from baseline to Month 4 in DPREMB-R or AAQoL-R were evaluated for PRC-063 versus LDX. The DPREMB-R is used to assess ADHD symptom severity over the past month by time of day and consists of 12 items that are grouped into two subscales, which includes morning (AM) and evening (PM) subscales. Three questions address specific morning behaviors and nine questions address specific evening behaviors. The DPREMB-R has been demonstrated to be a reliable and valid measure of the effect of ADHD symptoms on the behavior of children (Faraone et al., 2018). A 3-point decrease in the AM scale and a 5-point decrease in the PM scale have been correlated to a Clinical Global Impressions (CGI)-Improvement Score of ≤2 (much or very much improved) (Wilens et al., 2022). Remission has been defined as ≤4 in the AM scale and ≤10 in the PM scale for children 8 years old or younger and ≤3 in the AM scale and ≤8 in the PM scale for children aged 9 years old or older (A. C. Childress et al., 2021). The AAQoL-R assesses the impact of the subject’s ADHD symptomatology on everyday activities and QoL over the past 2 weeks (Brod et al., 2006). With permission from the author, the scale was modified so that each item was rated in specific regard toward evening behavior. An approximate 8-point increase in the total score of the AAQoL from baseline has been proposed as the MCID (Tanaka et al., 2019).
The PSS was completed at baseline through Month 4. The secondary endpoint of change from baseline to Month 4 in PSS was collected for PRC-063 subgroups and the change from baseline to Month 4 in PSS PRC-063 versus LDX was evaluated. The PSS assesses subject experience on ADHD treatments, with questions on sleep, appetite, duration of action, and overall satisfaction with the ADHD treatment.
Safety Assessments
Safety assessments included reported and non-directed spontaneous AEs. Association between the event and study treatment was assigned by the Investigator. Subjects were monitored as per standard of care with regard to warnings and contraindications in the Product Monographs for PRC-063 and LDX. Blood pressure, temperature, pulse, and respiratory rate were assessed at baseline through Month 4.
Statistical Analysis
Demographics, patient characteristics, and primary and secondary endpoints were analyzed using the efficacy population, defined as the enrolled population who had taken ≥1 dose of the study medication. The primary endpoint was analyzed separately for pediatric and adult subjects by treatment group using a paired t-test. The key secondary outcome was a non-inferiority comparison of the change in the ADHD-5-RS total score from baseline to Month 4 between subjects who received PRC-063 and those who received LDX, analyzed separately for adult and pediatric subjects using a generalized linear model (GLM). The analysis was dependent on the upper limit of a two-sided confidence interval (CI) of the observed treatment difference between PRC-063 and LDX. To establish non-inferiority, the upper limit between PRC-063 and LDX needed to be less than 6.6 (the MCID for the ADHD-5-RS) (Storebø et al., 2015; Zhang et al., 2005). For the non-inferiority test using a one-sided two-sample equal-variance t-test, it was assumed that the treatment difference was 0 (i.e., no treatment difference) and the standard deviation of the treatment difference was 13 for the pediatric group and 11 for the adult group. With a sample size of 98 pediatric patients (46 PRC-063-treated and 46 LDX-treated) and a non-inferiority margin of 6.6, the study had 80% power to demonstrate non-inferiority of PRC-063 as compared to LDX at the significance level of 0.05. With a sample size of 72 adult patients (36 PRC-063-treated and 36 LDX-treated) and a non-inferiority margin of 6.6, the study had 81% power to demonstrate non-inferiority of PRC-063 as compared to LDX at the significance level of 0.05. To control for the observed baseline differences between treatment groups, a post-hoc propensity score was estimated for each of the adult and pediatric populations using variables for study site, age, gender, currently receiving treatment for ADHD, and baseline ADHD-5-RS Total Score.
A similar statistical approach was used to analyze the change in WFIRS-P and DPREMB-R scores from baseline to Month 4 for pediatric subjects who received PRC-063 and the change in WFIRS-S and AAQoL-R scores from baseline to Month 4 for adult subjects who received PRC-063. The change in these scores were analyzed using two sample t-tests. The change in PSS survey responses was analyzed for pediatric and adult subjects by treatment group using a chi-squared test. A post-hoc analysis of the WFIRS-P, WFIRS-S, AAQoL-R and DPREMBR scores compared the rates of improvement based on MCID and remission, where available, for both treatments.
Due to the COVID-19 pandemic, Protocol Administrative Clarifications were submitted and approved by ethics committees during the conduct of the study to manage study participants due to investigator site closures or restrictions placed for inpatient visits. In brief, follow-up visits were completed virtually (if feasible and permissible) per the site’s discretion, out of window visits were permissible, missing assessments for height and weight were documented in the patients’ source documents and in the CRFs as not completed, and patient questionnaires were completed virtually. Protocol deviations were not issued for missed assessments and out of window visits.
Continuous variables were reported as the number of observations, number of missing observations, mean, standard deviation (SD), median, first quartile, third quartile, and minimum and maximum. Categorical variables were presented as the number and percentage of subjects in each category. Statistical testing was performed at the two-sided .05 significance level unless otherwise specified and p-values were not adjusted for multiple comparisons.
Treatment-emergent adverse events (TEAEs) were categorized by system organ class and preferred term coded according to the MedDRA dictionary (version 24.0). TEAE summaries were based on the number of subjects experiencing an event, not the number of AEs experienced. Subjects who reported more than one instance of the same AE are only reported once in the TEAE summary. The denominator used for calculation of the percentages was the number of subjects in the Safety population (i.e., the enrolled population who had taken at least one dose of study medication) in each treatment group.
Results
Subject Disposition
A total of 257 subjects were screened and of these subjects, 255 subjects were enrolled. In the pediatric population, 74 patients were assigned to PRC-063, 66 patients took at least one dose of study medication and 56 patients completed all study visits while 18 (24.3%) patients withdrew and 69 patients were assigned to LDX, 68 patients took at least one dose of study medication and 55 patients completed all study visits in the LDX group while 14 (20.3%) patients withdrew. The most common reasons for discontinuation in the pediatric population (including those who did not take study medication) were withdrawal of consent (PRC-063: 7, LDX: 2), lost to follow-up (PRC-063: 2; LDX: 0), and AEs (PRC-063: 7; LDX: 10). In the adult population, 58 patients were assigned to PRC-063, 46 took at least one dose of study medication and 42 patients completed all study visits while 16 (27.6%) patients withdrew and 54 patients were assigned to LDX, 46 took at least one dose of study medication and 43 patients completed all study visits in the LDX group while 11 (20.4%) patients withdrew. The most common reasons for discontinuation in the adult population (including those who did not take study medication) were withdrawal of consent (PRC-063: 0; LDX: 2), lost to follow up (PRC-063: 5, LDX: 1), and AEs (PRC-063: 9, LDX: 6).
Subject Demographics and Characteristics
In Table 1, subject demographics and characteristics are presented for the adult and pediatric population.
Subject Demographics and Characteristics.
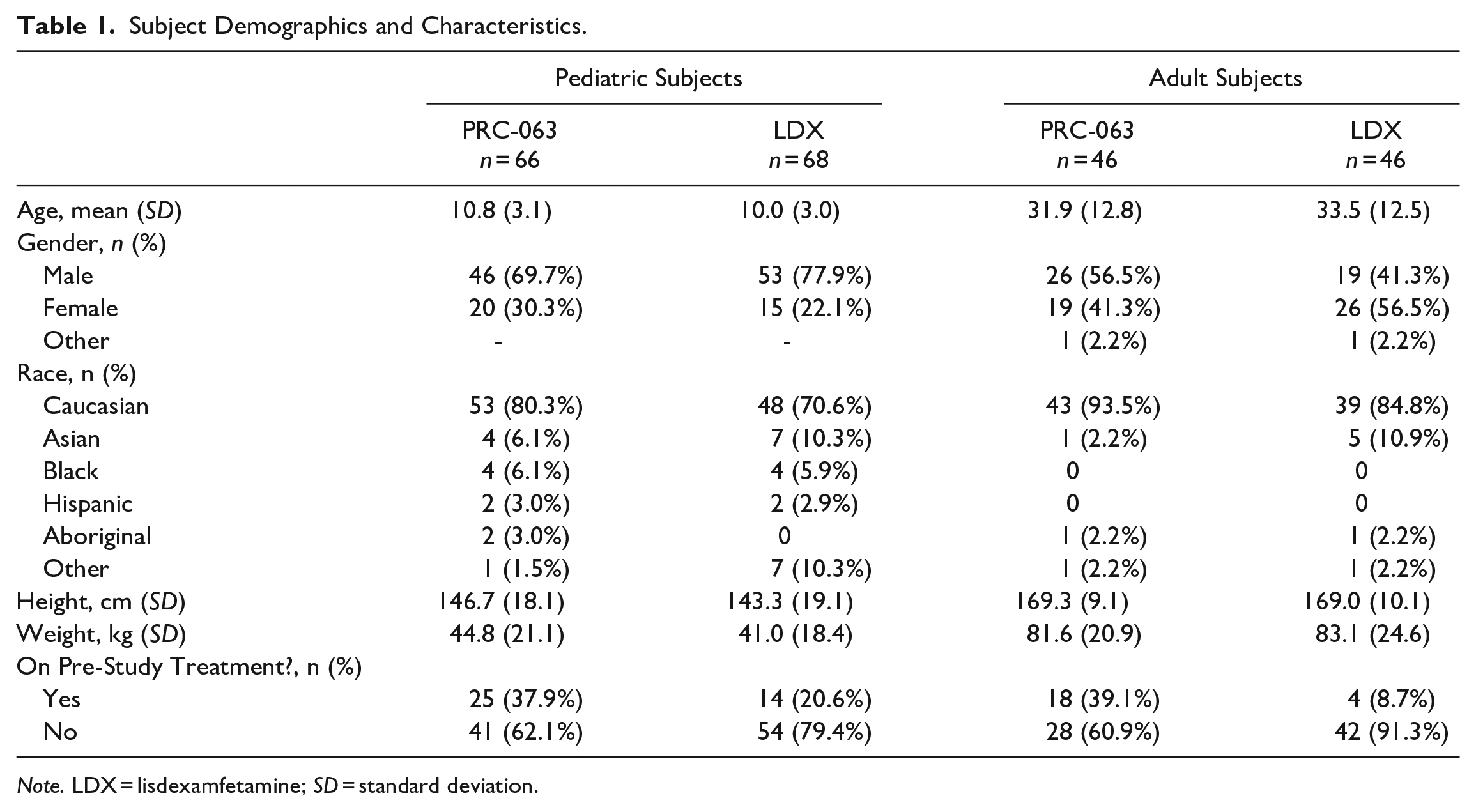
Note. LDX = lisdexamfetamine; SD = standard deviation.
In the pediatric population, the mean (SD) age was 10.8 (3.1) years for PRC-063 and 10.0 (3.0) years for LDX subjects. The majority of PRC-063 (46/66, 69.7%) and LDX (53/68, 77.9%) subjects were male and most were Caucasian (PRC-063: 80.3% and LDX: 70.6%). The majority of PRC-063 (62.1%) and LDX (79.4%) subjects were not currently receiving treatment for ADHD at the time of study enrollment (i.e., treatment-naïve; patients who were not receiving pharmacotherapy for the treatment of ADHD). While the direction of the distribution for each patient characteristic were similar between treatments, between-treatment differences in the ratios of each measure were observed. The most common comorbidities reported in the pediatric group were anxiety (PRC-063: 24/66, 36.4% and LDX: 24/68, 35.3%), oppositional defiant disorder (PRC-063: 15/66, 22.7% and LDX: 11/68, 16.2%), autism spectrum disorder (PRC-063: 9/66, 13.6% and LDX: 5/68, 7.4%), learning disorder (PRC-063: 8/66, 12.1% and LDX: 5/68, 7.4%), tic disorder/Tourette’s syndrome (PRC-063: 6/66, 9.1% and LDX: 6/68, 8.8%) and asthma (PRC-063: 4/66, 6.1% and LDX: 6/68, 8.8%).
In the adult population, the mean (SD) age was 31.9 (12.8) years for PRC-063 and 33.5 (12.5) years for LDX subjects. More than half of PRC-063 subjects were male (26/46, 56.5%) and the majority of LDX subjects were female (26/46, 56.5%). Most of the adult subjects were Caucasian (PRC-063: 93.5% and LDX: 84.8%). The majority of PRC-063 (60.9%) subjects were not currently receiving treatment for ADHD at the time of study enrollment (i.e., treatment-naïve), and a greater proportion of LDX subjects were treatment naïve (91.3%). Differences in the distribution of male-to-female ratios and the proportion of treatment-naïve patients were observed in this patient population. The most common comorbidities reported in the adult group were anxiety (PRC-063: 18/46, 39.1% and LDX: 19/46, 41.3%), depression (PRC-063: 16/46, 34.8% and LDX: 12/46, 26.0%), insomnia (PRC-063: 7/46, 15.2% and LDX: 4/46, 8.7%) and asthma (PRC-063: 8/46, 17.4% and LDX: 8/46, 17.4%).
Dose-optimization
The distributions of optimized study medication dose (mg/day) for subjects treated with PRC-063 and LDX in the Safety population are presented in Figure 1A and B, respectively. In pediatric subjects, the optimized daily dose of PRC-063 ranged from 12.5 to 85 mg/day with most subjects optimized between 35 and 70 mg/day (mean ± SD: 49.8 ± 14.9 mg/day or 1.25 ± 0.57 mg/kg/day). Two pediatric patients were administered off-label doses of PRC-063 (1 received 12.5 mg and 1 received 85 mg). The optimized daily dose for adult subjects for PRC-063 ranged from 25 to 125 mg/day with most subjects optimized at 35 to 70 mg/day (mean ± SD: 57.3 ± 18.7 mg/day or 0.70 ± 0.27 mg/kg/day). One adult patient was administered an off-label dose of PRC-063 (1 received 125 mg). In the pediatric population, LDX-optimized doses ranged from 10 to 60 mg/day with most subjects optimized at 30 to 60 mg/day (Mean ± SD: 29.2 ± 14.6 mg/day or 0.75 ± 0.41 mg/kg/day). For adults treated with LDX, optimized daily doses ranged from 15 to 70 mg/day, with most subjects optimized at 30 to 50 mg/day (Mean ± SD: 39.5 ± 13.4 mg/day or 0.48 ± 0.19 mg/kg/day). Two adult patients were administered off-label doses of LDX (two received 70 mg).
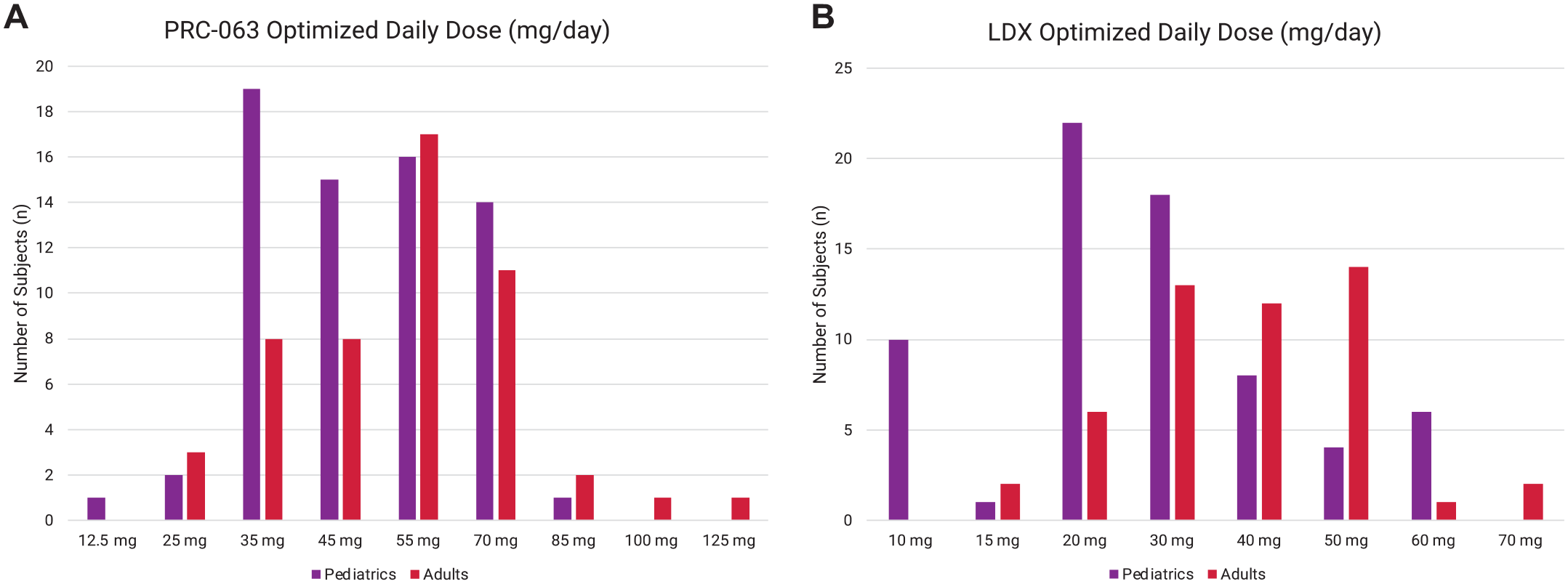
Distribution of optimized (A) PRC-063 and (B) LDX dosages for pediatric and adult safety populations.
Efficacy
ADHD-5-RS Total Score Change From Baseline
PRC-063 was effective in reducing ADHD symptoms in pediatric and adult subjects. The primary efficacy endpoint, change from baseline in ADHD-5-RS total score, was statistically significant (p < .001) after a 4-month period of treatment for both populations. The mean (SD) ADHD-5-RS total score change from baseline at Month 4 for pediatric subjects treated with PRC-063 was −16.6 (10.4) and ranged from −37.0 to 1.0 (Figure 2A). The mean (SD) ADHD-5-RS total score change from baseline at Month 4 for adult subjects treated with PRC-063 was −14.8 (10.6) and ranged from −35.0 to 12.0 (Figure 2B).
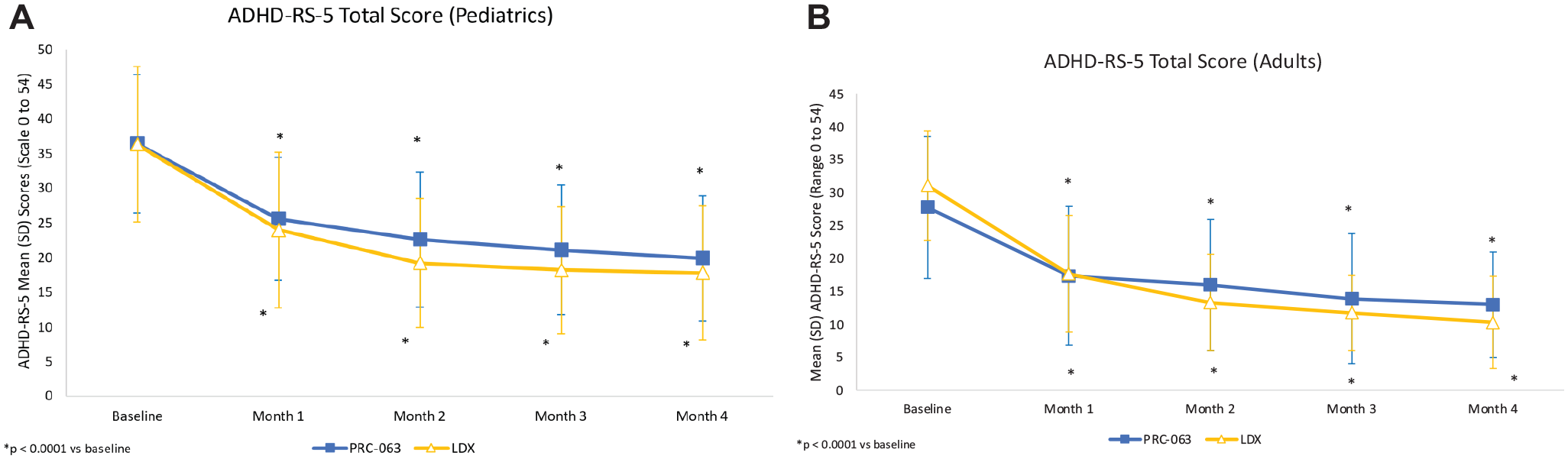
Mean (SD) ADHD-5-RS scores at all study visits in (A) pediatric and (B) adult subjects with ADHD treated with PRC-063 versus LDX.
The key secondary efficacy analysis, the ADHD-5-RS total score change from baseline to Month 4, was analyzed for the adult and pediatric populations for the efficacy and PP analyses. In the pediatric efficacy and PP analyses, the least square (LS) mean treatment difference (standard error [SE]; 95% confidence interval [CI]) was 1.68 (1.70; 95% CI [−1.70, 5.06]) and 1.69 (1.72; 95% CI [−1.73, 5.10]), respectively. In the adult population, the LS mean treatment difference (SE; 95% CI) was 4.40 (1.98; 95% CI [0.45, 8.35]) in the efficacy population and 4.38 (1.98; 95% CI [0.42, 8.34]) in the PP population.
The upper limit of the 95% CI of the observed treatment difference was greater than 6.6 in the adult population, and therefore PRC-063 failed to meet non-inferiority criteria to LDX (Figure 3). In the pediatric population, the observed treatment difference was less than 6.6; therefore, PRC-063 was considered non-inferior to LDX (Figure 3). However, when a propensity score analysis was conducted to control for baseline differences between groups, PRC-063 remained non-inferior to LDX in the pediatric population (LS mean difference [SE]: 1.772 [2.314]; 95% CI [−2.818 to 6.362]), while the comparison in the adult population was inconclusive (LS mean difference [SE]: 4.569 [2.997]; 95% CI [−1.396 to 10.533]), crossing both the 0 and the 6.6.boundaries.
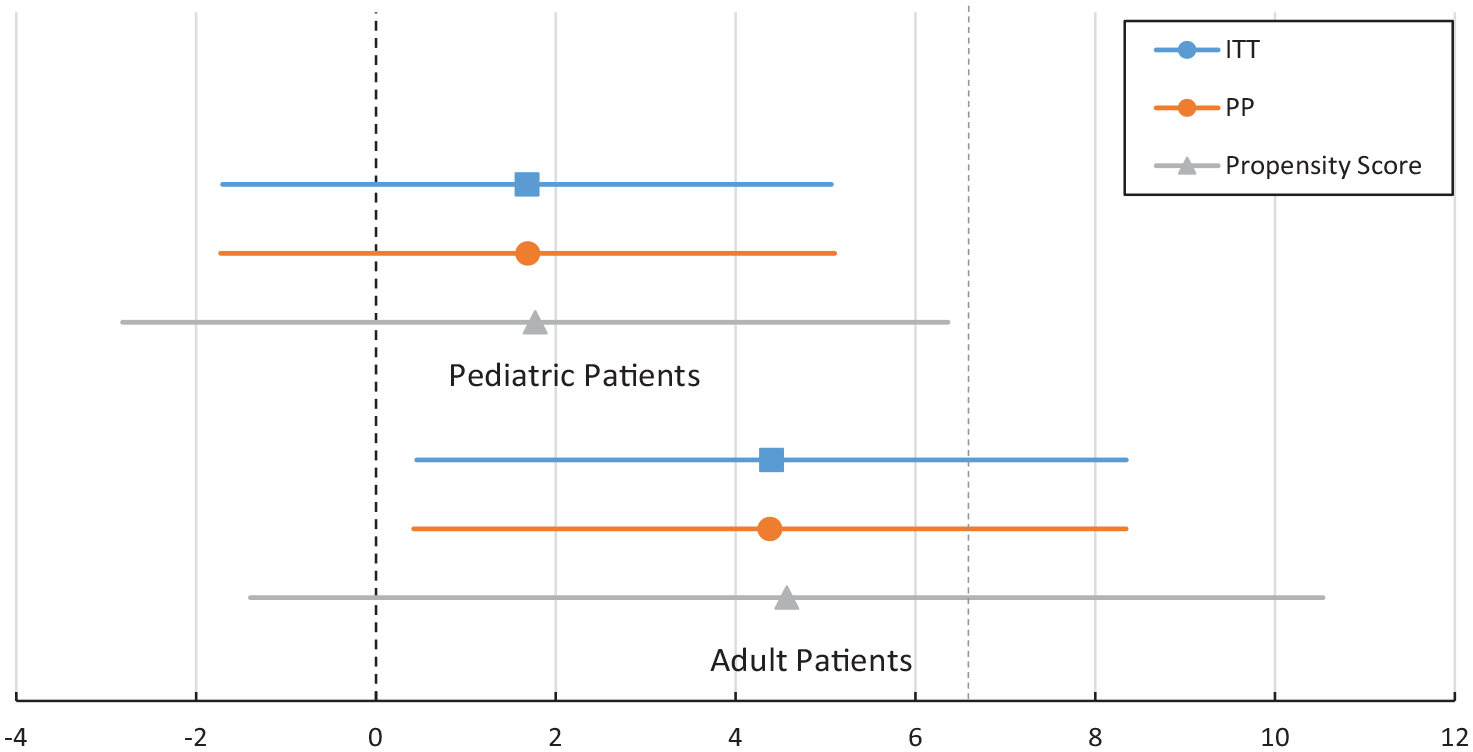
Non-inferiority analysis of between treatment difference in ADHD-5-RS total score for PRC-063 versus LDX in the ITT and PP adult and pediatric populations. Propensity score analysis is included in gray.
WFIR-P/-S Total Score Change From Baseline
Improvements in functional impairments were demonstrated after a 4-month period in the pediatric and adult populations following treatment with PRC-063. A statistically significant mean (SD) WFIRS total score change from baseline was demonstrated at Month 4 in pediatric subjects (PRC-063: −0.36 [0.41]; LDX:_−0.25 [0.31], p < .0001) (Figure 4A) and adult subjects (PRC-063: −0.54 [0.36]; LDX: −0.66 [0.44], p < .0001) treated with PRC-063 (Figure 4B). For both treatment groups and in both age populations, significant improvements from baseline were demonstrated by Month 1 and continued to Months 2, 3, and 4 (p < .0001 vs. baseline). However, there was no significant between-treatment differences in mean (SD) WFIRS total score change from baseline at Month 4 for adult subjects (p = .2802) or for pediatric subjects (p = .1782). There were also no significant between-treatment differences in any WFIRS subscales from baseline to Month 4. When improvements on the WFIRS-S and WFIRS-P were stratified by those who achieved the MCID or greater (≥0.25) versus those who did not, 60.0% of pediatric patients receiving PRC-063 achieved a MCID or greater on the WFIRS total score versus 50.0% of pediatric patients receiving LDX. Of adult patients, 77.3% receiving PRC-063 achieved a MCID or greater on the WFIRS total score versus 81.4% receiving LDX.
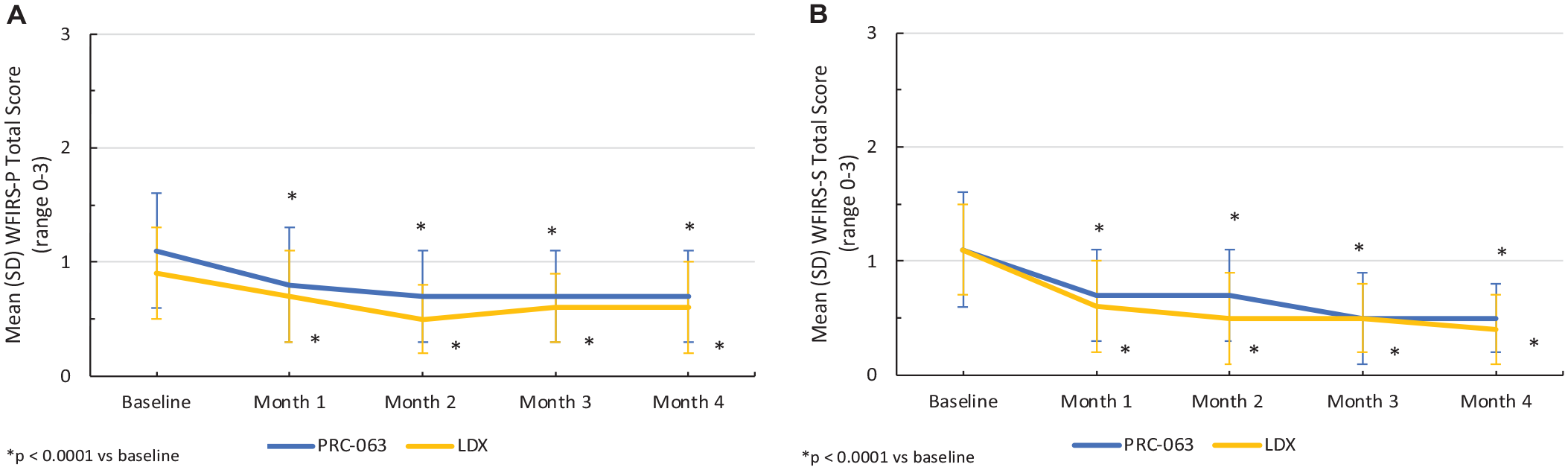
Mean (SD) WFIRS scores at all study visits in (A) pediatric (WFIRS-P) and (B) adult (WFIRS-S) subjects with ADHD treated with PRC-063 versus LDX.
DPREMB-R
The mean (SD) DPREMB-R AM score change from baseline at Month 4 for pediatric subjects treated with PRC-063 was −1.5 (2.2) (p < .0001). For PRC-063 treatment, significant improvements were demonstrated from baseline to Month 1 and continued to Months 2, 3, and 4 (p < .05 vs. baseline), LDX treatment demonstrated significant improvements from baseline to Months 1 and 2 (p < .05 vs. baseline), but not Months 3 and 4 (LDX: −0.8 [3.0], p > .05 vs. baseline). Improvements in the AM score were greater than or equal to the MCID in 27.4% of patients receiving PRC-063 and 29.6% of patients receiving LDX. Remission levels in the AM score were achieved in 74.2% of patients receiving PRC-063 and 72.2% of patients receiving LDX as measured at end of study. The improvement in morning behavior was not significantly different between treatments at Month 4 (p = .2458).
There was a significant improvement in evening behavior in the pediatric population who were treated with PRC-063. The mean (SD) DPREMB-R PM score change from baseline at Month 4 for pediatric subjects treated with PRC-063 was −5.7 (4.9) (p < .0001). For both treatment groups, significant improvements were demonstrated from baseline to Month 1 and continued to Months 2, 3, and 4 (p < .0001 vs. baseline); however, the improvement in evening behavior was significantly greater in the PRC-063 group compared to the LDX group (3.6 [5.7], p = .0479) (Figure 5). Improvements in the PM score were greater than or equal to the MCID in 58.0% of patients receiving PRC-063 and 46.3% of patients receiving LDX. Remission levels in the PM score were achieved in 60.7% of patients receiving PRC-063 and 56.6% of patients receiving LDX as measured at end of study.
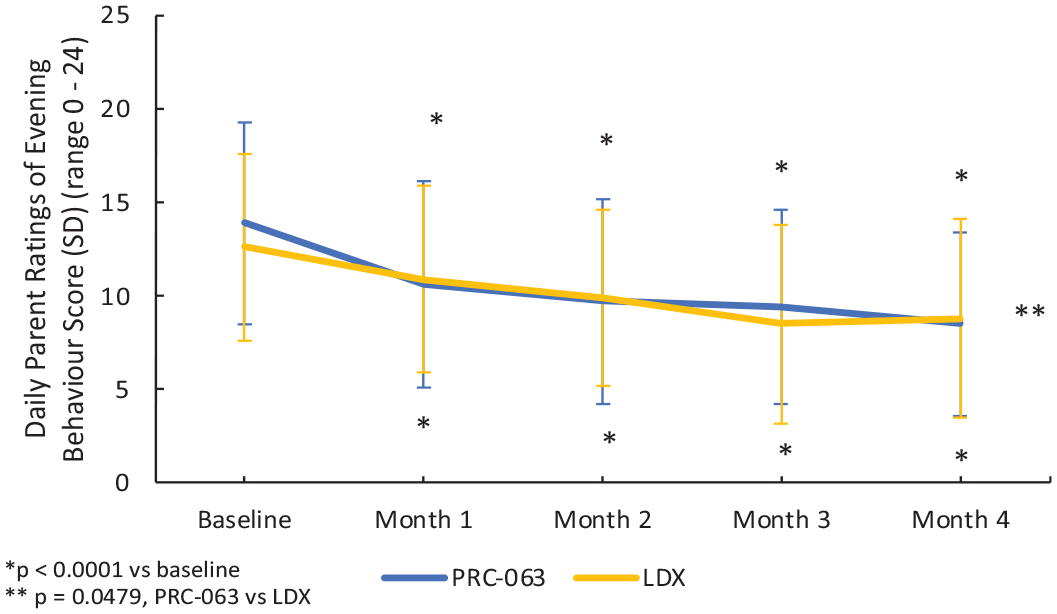
Mean (SD) DPREMB-Evening Behavior scores all study visits in pediatric subjects treated with PRC-063 versus LDX.
AAQoL-Revised
There was a significant improvement in evening QoL in adult subjects treated with PRC-063 after a 4-month period. The mean (SD) AAQoL-R total score change from baseline at Month 4 for adult subjects treated with PRC-063 was 20.3 (13.7) (p < .001). For both treatment groups, significant improvements were demonstrated from baseline to Month 1 and continued to Months 2, 3, and 4 (p < .0001 vs. baseline). The mean (SD) AAQoL-R total score change for LDX subjects was 25.2 (15.6); there was no significant difference between PRC-063 and LDX treatment groups (Figure 6). When improvements on the AAQoL-R were stratified by those who achieved the MCID or greater (≥8 improvement) versus those who did not, 79.6% of adult patients receiving PRC-063 achieved the MCID or greater versus 84.0% of adult patients receiving LDX.
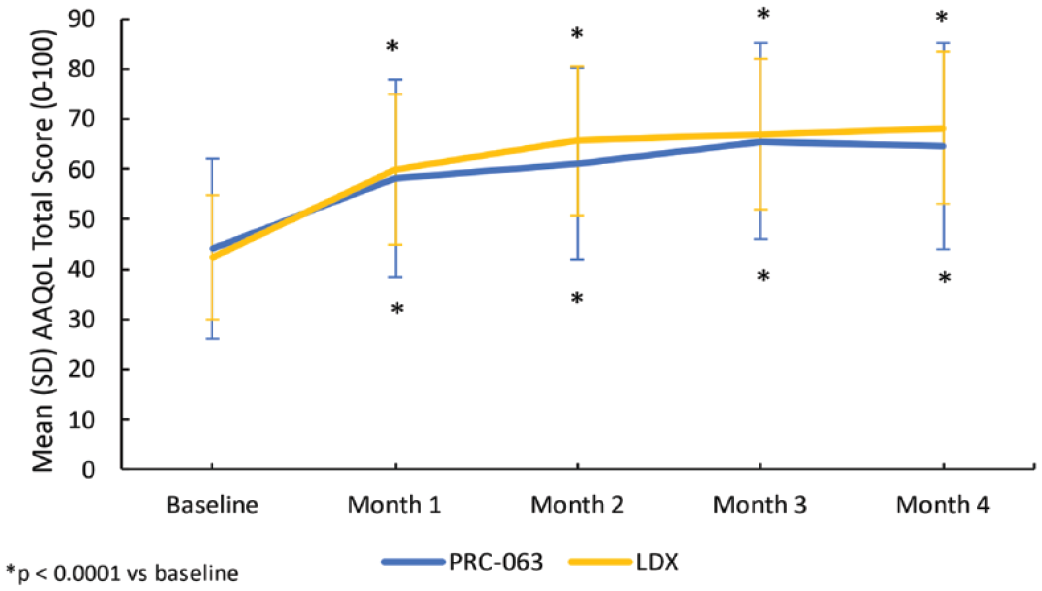
Mean (SD) AAQoL total score (0–100) at all study visits in adult subjects treated with PRC-063 versus LDX.
PSS
Results of the PSS in pediatric and adult populations for all subjects are presented in Supplemental Appendix Figure 1. In the adult population, the only between-treatment statistical difference demonstrated was in evening symptom control (as assessed by Question 4: When taking your ADHD/study medication, how satisfied are you with you ADHD symptoms in the evening?) between the two non-naïve treatment groups (i.e., subjects who were receiving psychotherapy for the treatment of ADHD at the time of study enrollment). Improvements from baseline to end of study were reported in 14 (87.5%) subjects treated with PRC-063 compared to zero patients treated with LDX (p = .0103). In the pediatric population, the only between-treatment statistical difference demonstrated was in lunch appetite (as assessed by Question 5: When taking your ADHD/study medication, how satisfied are you with your level of appetite at lunch?) between the two non-naïve treatment groups. Improvements in lunchtime appetite from baseline to end of study were reported in 16 (84.2%) subjects treated with PRC-063 versus 3 (33.3%) LDX-treated subjects (p = .0127).
Safety
Treatment-emergent adverse events (TEAEs) that occurred in ≥5% of the safety population for either pediatric or adult subjects are presented in Table 2.
TEAEs Occurring in ≥5% of the Adult and Pediatric Safety Populations.
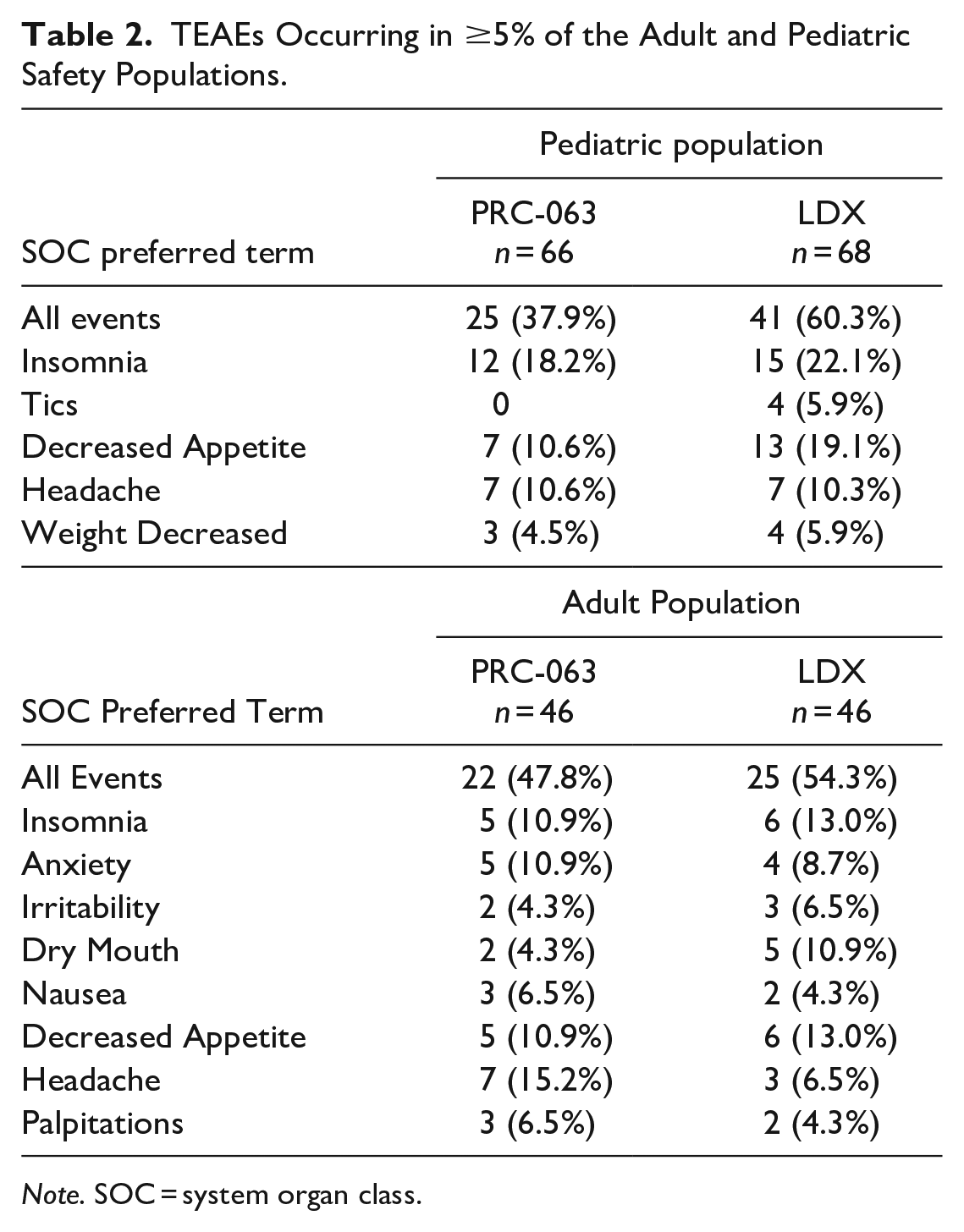
Note. SOC = system organ class.
In the pediatric population, 37.9% (25/66) subjects in the PRC-063 group reported 72 TEAEs (mild = 48, moderate = 23, and severe = 1) compared to 60.3% (41/68) subjects in the LDX group reported 110 TEAEs (mild = 79, moderate = 30, and severe = 1). A higher percentage of TEAEs that led to study discontinuation occurred in the LDX group versus the PRC-063 group (17.6% [12/68] vs. 6.1% [4/66]).
In the adult population, 47.8% (22/46) subjects in the PRC-063 group reported 76 TEAEs (mild = 34, moderate = 40, and severe = 2) and 54.3% (25/46) subjects in the LDX group reported 79 TEAEs (mild = 22, moderate = 37, and severe = 20). No serious AEs were reported. TEAEs that led to study discontinuation occurred in 2.2% (1/46) subjects in the PRC-063 group and 4.3% (2/46) subjects in the LDX group.
Discussion
In this prospective, RWE study of patient- and clinician-reported outcomes conducted over a 4-month period across Canada and including subjects across the ADHD lifespan, treatment with PRC-063 and LDX resulted in a significant reduction in ADHD symptoms, improved evening behavior and QoL, and improved functionality. Following 4 months of treatment, with monthly follow-up visits, ADHD symptomatology reduced by 45.6% in the pediatric group receiving PRC-063 and by 53.2% in the adult group receiving PRC-063. These improvements were more than twice the minimal MCID of the scale used to measure the change. This study supports the findings of the existing RCT evidence base for PRC-063 (A. C. Childress et al., 2020; A. Childress et al., 2022; Weiss, Childress, & Donnelly, 2021; Weiss, Cutler et al., 2021; Weiss, Surman et al., 2021; S. B. Wigal et al., 2020) and LDX (Adler et al., 2008; T. Wigal et al., 2010) and adds to a greater understanding of the comparative efficacy and safety of two commonly used, currently available ADHD treatment options in Canadian clinical practice.
Non-inferiority of PRC-063 was established with LDX for the pediatric population, but not the adult population. When the observed differences in baseline values were accounted for using a propensity score analysis for potential confounders, non-inferiority was not demonstrated for the adult population (results were inconclusive, as the 95% confidence intervals straddled both MCID and 0 boundaries), but results remained non-inferior for the pediatric population. Morning behaviors at the end of treatment, as measured with the DPREMB-R, were significantly improved from baseline with PRC-063 treatment, but not LDX treatment, although between-treatment differences were not statistically significant. When evening behaviors were rated using the DPREMB-R in the pediatric population and the AAQoL-Revised in the adult population, both treatments demonstrated statistically significant improvements from baseline and the PRC-063 group demonstrated greater improvements than the LDX group in the pediatric population, although no between-treatment differences were found in the adult population. In addition, measurements of functional impairment using the WFIRS demonstrated improvement greater than the MCID over 4 months of treatment in both PRC-063 and LDX treatment groups, although no between-treatment differences were observed.
Non-inferiority between treatments is supported by a previously-conducted, double-blind, randomized, non-inferiority crossover comparison of PRC-063 and LDX on driving performance measured in a simulator in adults (NCT02555150) which demonstrated no difference between treatments. The optimized doses in that blinded crossover study were 73.52 ± 17.87 mg/day of PRC-063 and 49.55 ± 11.80 mg/day of LDX.
In this real-world, non-randomized study, the mean (SD) optimized daily dose for adult subjects treated with PRC-063 was 57.3 ± 18.7 mg/day. Notably, the mean dosage was approximately 15 mg/day lower than the optimized dosage that was observed in a 6-month dose-optimized open-label extension study of PRC-063 in adults (73.3 ± 21.8 mg/day; NCT02168127) (Weiss, Childress, & Donnelly, 2021), as well as the previously-mentioned driving performance study. In the current study, the mean (SD) optimized daily dose for LDX-treated adults subjects was 39.5 ± 13.4 mg/day, lower than that in the previously-mentioned driving performance study as well as in an open-label, randomized, crossover efficacy and safety study designed of LDX in adults with ADHD (T. Wigal et al., 2010). It is therefore possible that some adult subjects were underdosed in both PRC-063 and LDX treatment groups, whether due to unfamiliarity with the treatment or a reluctance to use the higher doses, contributing to the inconclusive result for the corrected non-inferiority propensity score analysis. Appropriate dose optimization was difficult to include in the propensity analysis, as optimal dose may differ from patient-to-patient, but the mean dose was lower than that what was predicted prior to the study. Further analysis is warranted to establish the relative efficacies of PRC-063 and LDX in a real-world adult population. Nonetheless, dose optimization remains an important consideration in achieving optimal outcomes for these ADHD therapies.
This real-world evidence study included several questionnaires designed to measure real world outcomes. With 4 months of treatment, in which statistically significant improvements in symptom scores were observed within 1 month of treatment, 60% and 50% of pediatric patients who respectively received optimized PRC-063 or LDX treatment demonstrated clinically meaningful improvements in functional outcomes. In adult patients, the clinically meaningful improvements in functionality were observed in even larger proportions (77% and 81% of patients who respectively received optimized PRC-063 or LDX treatment). These results are supported by the observations of a QoL tool that specifically examines evening measures in adults, in which clinically significant improvements were measured in 79.6% of patients receiving PRC-063 and 84.0% of patients receiving LDX. Likewise, measurements of time of day behaviors in children met remission levels for morning measures and evening measures in, respectively, 74.2% and 60.7% of patients receiving PRC-063 and 72.2% and 56.6% of patients receiving LDX.
The current study provides insight into the tolerability of long-acting stimulants in Canadian clinical practice. In the CADDRA (2020) guidelines, both MPH and amphetamines are recommended as first-line treatment options in ADHD; however, the guidelines state that patients may tolerate or respond to one class better than the other. In the current RWE study of two commonly used ADHD therapies, most AEs reported were of mild or moderate severity and no serious AEs were reported. Although the data suggests a trend in differences in tolerability between PRC-063 and LDX in the safety population, as this was a non-randomized observational study, this trend may be due to other, non-controlled factors. In the pediatric safety population, AEs that led to discontinuations were higher in the group receiving LDX than that receiving PRC-063 (17.6% vs. 6.1%), as well as the number of patients reporting any AE (60.3% vs. 37.9%) and the number of events reported by patients (110 vs. 72 events). Of the AEs most commonly associated with stimulant therapy, headache was similar between treatments in the pediatric population, but insomnia and decreased appetite were higher in the LDX treated group. These observations suggest that PRC-063 may be better tolerated than LDX as an ADHD medication in pediatric subjects, while demonstrating similar levels of efficacy. In the adult population, AEs that led to discontinuations were similar in LDX and PRC-063 groups (4.3% vs. 2.2%) with slightly higher reports of AEs in the LDX group (54.3% of LDX patients reporting 79 events vs. 47.8% of PRC-063 patients reporting 76 events). The severity of events was also higher in the LDX group (20 severe events vs. 2 in the PRC-063). Of the AEs most commonly associated with stimulant therapy, headache was higher in the PRC-063 treated group in the adult population, but dry mouth, insomnia, and decreased appetite were higher in the LDX treated group (Table 2).
Post-hoc analyses of phase 3 studies with both LDX and MPH arms have reported conflicting results (D. R. Coghill et al., 2014; Soutullo et al., 2013). Soutullo et al. (2013) demonstrated a 5.6 difference in ADHD-Rating Scale score in favor of LDX, although the mean ± SD optimized doses were 53.8 ± 15.6 mg/day for LDX and 45.4 ± 12.7 mg/day for OROS-MPH, as reported in the original manuscript (D. Coghill et al., 2013), suggesting the methylphenidate arm may have been underdosed. A similar study by Coghill and colleagues found no statistically significant differences between treatments (D. R. Coghill et al., 2014). Of note, patients who received MPH demonstrated less improvement if they were previously on treatment compared to those who were treatment naïve. Meta-analyses examining safety and tolerability of ADHD therapies report equivocal findings. Results of a meta-analysis that examined the efficacy and safety of ADHD therapies (i.e., guanfacine, atomoxetine, LDX, and MPH) in children and adolescents, including 36 RCTs, showed a similar finding to our study–immediate-release MPH was the most tolerable among all therapies and the least likely to be discontinued (Joseph et al., 2014). In the most comprehensive network meta-analysis to-date including 133 double-blind RCTs, Cortese et al. (2018), examined the efficacy and tolerability of amphetamines (including LDX), atomoxetine, bupropion, clonidine, guanfacine, MPH, and modafinil in children, adolescents, and adults with ADHD. No substantial differences were observed in terms of efficacy, and no differences in tolerability were demonstrated between medications. In this meta-analysis, the authors also found no substantial differences in efficacy or tolerability when the maximum dose, as defined by FDA or by guidelines, was used. The results of this RWE study found non-inferiority between treatments in children and adolescents and were inconclusive regarding adults, whereas the meta-analysis authors recommended use of MPH in children and adolescents and amphetamines in adults.
There were several unanticipated baseline treatment differences observed between treatment groups in this study. In the pediatric population, baseline ADHD-5-RS scores were well-matched; however, in the adult population, the subjects treated with PRC-063 had lower scores versus LDX at baseline, providing less variance for change. Since this study was not blinded, it was observed that investigators put more severe pediatric patients in the LDX treatment group. In the pediatric population there were substantially more males than females, which is not surprising given ADHD is more commonly diagnosed in youth in males than females (Mowlem et al., 2019). In both the pediatric and particularly the adult populations, a higher percentage of subjects were receiving pre-study treatment in the PRC-063 groups versus the LDX groups. It is conceivable that investigators in this study were more likely to allocate adult patients not currently receiving treatment at the time of enrollment into the LDX treatment group.
Limitations of the study are those inherent to the design of open-label studies, which have potential for bias because treatments are non-randomized and unblinded. One potential weakness of a RWE study are the potential unobserved factors that may influence the investigator’s treatment decisions. Physician opinion, patient request, and awareness of a clinical trial (i.e., patients enrolling in a clinical trial may be different to those in the general population; physicians may be more likely to try a novel treatment on a healthier or safer population) (Unger et al., 2019) are three factors identified by Beaulieu-Jones et al. (2020) as potentially affecting the results of a RWE study. The possibility of these biases make differentiation between the effects of patient, treatment, and physician difficult. In this study, there were baseline differences in subjects that might have contributed to the results. In particular, the LDX adult group had approximately 30% more treatment-naïve patients (for a total of 91.3%) than the PRC-063 group, making head-to-head comparison problematic. Moreover, there was a potential for bias based on prior experience with therapy (e.g., PRC-063 was approved more recently, while LDX is well-established), name recognition, and preference for newer therapies. Despite these limitations, the study examined real-world data and did not limit who could be enrolled beyond investigator decision of appropriateness for study participation, the ability to sign informed consent, attend visits, and complete rating scales while meeting the approved labeling for each product, which is more reflective of community practices than RCTs. Differences between treatment groups, in addition to providing efficacy and safety information, may therefore also indicate physician or patient preference.
Conclusion
This real-world study adds to the current RCT evidence base on the efficacy and safety of PRC-063 and LDX and provides additional insight into outcomes not previously examined in RCTs. Both LDX and PRC-063 resulted in symptom improvements, and improvements in functionality in ADHD subjects were demonstrated across the lifespan. Both therapies were well-tolerated; however, a trend was observed toward a higher percentage of LDX pediatric subjects experiencing TEAEs compared to PRC-063 subjects. Dose optimization remains an important consideration in achieving the best outcomes for both medications.
Supplemental Material
sj-docx-1-jad-10.1177_10870547231172767 – Supplemental material for Real-World Efficacy and Safety of Extended-Release Methylphenidate (PRC-063) in the Treatment of ADHD in Pediatric and Adult Subjects: Results of a Phase IV Multicenter Comparison With Lisdexamfetamine Dimesylate
Supplemental material, sj-docx-1-jad-10.1177_10870547231172767 for Real-World Efficacy and Safety of Extended-Release Methylphenidate (PRC-063) in the Treatment of ADHD in Pediatric and Adult Subjects: Results of a Phase IV Multicenter Comparison With Lisdexamfetamine Dimesylate by Judy van Stralen, Gurdeep Parhar, Anita Parhar, Valérie Tourjman, Sohail Khattak, Tahira Ahmed, Graeme A. E. Donnelly and Jodan Ratz in Journal of Attention Disorders
Footnotes
Acknowledgements
Medical writing services were provided by Susan Bartko-Winters, PhD, of SBW Medical Writing Inc, which was funded by Elvium Life Sciences. We thank the reFOQus study group including Dr. Ahmed, Dr. Belle-Isle, Dr. Chandrasena, Dr. Chouinard, Dr. Kao, Dr. Khattak, Dr. Kjernisted, Dr. Ortega, Dr. Parhar, Dr. Robitaille, Dr. Shivakumar, Dr. Tourjman, and Dr. van Stralen.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: GAD and JR are employees of Elvium Life Sciences. JVS has received research support, consulting fees, and speaker fees from Elvium Life Sciences. GP has received honorarium, research support, and served on advisory boards for Elvium Life Sciences, Takeda, and Janssen. VT has received research support from Elvium Life Sciences. AP has received research support from Elvium Life Sciences. TA has received research support from Elvium Life Sciences.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Elvium Life Sciences. Medical writing services were funded by Elvium Life Sciences.
Supplemental Material
Supplemental material for this article is available online.
Author Biographies
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.