
Abstract
Introduction
ADHD is associated with significant functional impairments in multiple settings, including home, school, or work. Often, these impairments are not limited to the work or school day, but may begin upon wakening with continuation until the participant goes to sleep (Biederman et al., 2006). For adequate symptom management, some adults may require an ADHD medication that, when taken in the morning, provides a rapid onset of action and lasts well into the evening hours, or the equivalent time frame for those participants who have shift work. In a recent survey of caregivers for ADHD children, the three highest ratings of symptom severity by time of day were “Early Morning Routine,” “Evening Homework Time,” and “Bed Time” (Sallee, 2015). A survey examination of a medicated group of children with ADHD found that longer acting medications showed inferiority to shorter acting medications during morning routines, but superiority during the afternoon ratings (Coghill et al., 2008). Routines at bedtime and late evening were not different between medications but both were worse than in non-ADHD children (Coghill et al., 2008). Therefore, while stimulant preparations used for treatment of ADHD have varying onsets of action and durations of effect, there is an unmet medical need for a long-acting ADHD medication that has a rapid onset of action (i.e., within 1 hr) to control symptoms during the morning with duration of action that lasts well into the evening (i.e., >12 hr) to control symptoms for evening routines. PRC-063, a novel prolonged-release racemic methylphenidate hydrochloride (MPH) formulation, is designed to provide efficacy for those who require ADHD symptom control in the morning, throughout the day, and well into the evening.
PRC-063 is designed to be taken orally once daily in the morning or upon awakening. Following administration of PRC-063, the outer layer of MPH (20%) is immediately released and rapidly absorbed following ingestion reaching an initial maximum peak plasma concentration at 1.6 hr. When a 100 mg dose was given to healthy adult volunteers, the Cmax at steady state of the initial peak was 12.5 ± 3.7 ng/mL (Quinn et al., 2016). A protective coating prevents the innermost controlled-release layer (80% of MPH) from releasing until the gastrointestinal environment reaches a pH of 6.8 or greater, resulting in a gradual ascending concentration and a second, higher peak at 12.5 hr. In the same steady-state pharmacokinetic study, the Cmax of the second peak was 15.5 ± 4.6 ng/mL. MPH plasma levels then slowly decline with residual levels of approximately 18% at 24 hr post-administration (3.9 ± 2.7 ng/mL). As a result, steady state is achieved by the third day of once-daily dosing. This biphasic pharmacokinetic profile is intended to provide both a rapidly attained, initial post-dose peak concentration upon administration and a subsequent, more prolonged peak later in the day.
The study was conducted in the simulated adult workplace environment (AWE) setting (Wigal & Wigal, 2006; Wigal et al., 2010). The simulated AWE is a controlled environment with structured activities provided throughout the day, designed to provoke behaviors associated with ADHD symptoms and yield quantifiable outcomes. It is designed to monitor and quantitatively assess response to medication in the performance of adults engaged in activities simulating those that occur during a typical workday. The AWE is a proven tool for measuring attention- and behavior-associated outcomes in ADHD participants (Wigal & Wigal, 2006; Wigal et al., 2010). The Permanent Product Measure of Performance (PERMP) is a validated, time-sensitive, skill-adjusted test consisting of simple math problems to be completed at multiple time points throughout the simulated AWE session, and is a robust, objective measure of the ability to initiate a task, self-monitor/stay on task, and complete written seatwork (Wigal & Wigal, 2006). The PERMP does not test for mathematical ability or the ability to learn math because the difficulty of problems is adjusted to the existing math skill level of each participant to ensure that each individual achieves high accuracy rates.
In this study, the dose of PRC-063 was increased weekly until an optimal clinical response was achieved. The starting dose was 25 mg PRC-063 with a maximum permitted dose of 100 mg. Once the individually determined optimal dose had been achieved, the participant was maintained on that dose of PRC-063 and received active medication on one AWE Day and placebo on the other AWE Day in a randomized, blinded, crossover fashion.
The purpose of this randomized, double-blind, crossover, placebo-controlled, optimized-dose study was to assess the clinical efficacy, time of onset, and time course of efficacy over 16 hr of PRC-063 compared with placebo in an AWE setting in adults diagnosed with ADHD.
Method
Study Conduct
This double-blind AWE study design was based on a previously published study (Wigal & Wigal, 2006) and was conducted at two sites in the United States from September 2014 through March 2015 (CT Identifier NCT02225639). The AWE consisted of a classroom (with a chalkboard, tables, and chairs for seating two groups of participants and one group of observers, appropriate lighting, a stopwatch, and labels for seating assignments), a dosing room, a game/video room, a quiet room, an observation area, a meal area, a vital sign collection area, restrooms, and a phlebotomy room. Each AWE session had between 10 and 18 participants, two “teachers,” and two raters. The study protocol was approved by Institutaional Review Board (IRB) Services (Aurora, Ontario) and the State Research Advisory Panel of California. All patients gave written informed consent prior to the performance of any study-related procedures. All study-related activities were performed in accordance with good clinical practice (GCP) guidelines as required by the following: Declaration of Helsinki, 1964 (“Recommendations Guiding Physicians in Biomedical Research Involving Human Patients”) and all its Amendments to this date concerning medical research in humans; International Conference on Harmonization of Pharmaceuticals for Human Use Guideline for Good Clinical Practice (European Medicines Agency, 2002); European Union (EU) Clinical Trials Directive 2001/20/EC on the regulation of clinical trials in the EU; and the implementation of GCP and in accordance with applicable local regulations. In addition, participating adults signed an Authorization of Release of Personal Health Information for Research Purposes form when giving informed consent for participation in the study.
Participants
Seventy-five participants were screened for entry into the study. Of these, 16 participants were screening failures, and a total of 59 males and females aged between 18 and 60 years who met Diagnostic and Statistical Manual of Mental Disorders (5th ed.; DSM-5; American Psychiatric Association [APA], 2013) criteria for a diagnosis of any of the three subtypes of ADHD were enrolled if they met defined study inclusion and exclusion criteria. During the study screening, medical and psychiatric history data were collected and the diagnosis was based on clinician assessment using the Conners’s Adult ADHD Diagnostic Interview by Diagnostic and Statistical Manual of Mental Disorders (4th ed.; DSM-IV; APA, 1994; CAADID; Epstein, Johnson, & Conners, 2001).
Each participant was required to have (a) a baseline, clinician completed ADHD DSM-5 Rating Scale (ADHD-5-RS; adapted from DSM-5; APA, 2013) score equal to or greater than 24, as assessed after 7-day washout; (b) dissatisfaction with current pharmacological therapy for ADHD unless naïve to pharmacological therapy; (c) age-appropriate intellectual functioning as determined by an IQ of ≥80 on the Kaufman Brief Intelligence Test–2 (KBIT-2; Kaufman & Kaufman, 2004); (d) negative serum β-human chorionic gonadotropin pregnancy test at screening for females of childbearing potential, and use of a reliable and approved method of birth control unless post-menopausal or surgically sterile; and (d) a successful swallow test of an empty 100 mg study capsule. To be included in the study, adults who were currently or previously on stimulant treatment could be identified as positive stimulant responders with an inadequate stimulant medication response with their current medication either due to a suboptimal response or inadequate onset or duration of action, but not adverse stimulant responders.
Key exclusion criteria were presence of a comorbid psychiatric condition with severe symptoms, or other medical condition that could confound assessments, pose a risk to the participant, or prohibit study completion. Other exclusion criteria were allergy, serious adverse reaction or non-responsiveness to previous MPH therapy, pregnancy, substance or alcohol abuse, serious cardiac problems that may place the participant at increased vulnerability (e.g., symptomatic cardiovascular disease, advanced arteriosclerosis, or structural cardiac abnormality), history of strokes, epilepsy or migraine headaches, use of guanethidine, pressor agents, monoamine oxidase inhibitors, coumarin anticoagulants, anticonvulsants (e.g., phenobarbital, phenytoin, primidone), phenylbutazone, tricyclic antidepressants (e.g., imipramine, desipramine), selective serotonin reuptake inhibitors (SSRIs) or herbal remedies, clinically significant laboratory and/or electrocardiographic (ECG) abnormalities, elevated blood pressure defined as any values above 89 mmHg diastolic or 139 mmHg systolic as assessed at screening, known family history of sudden cardiac death or ventricular arrhythmia, currently considered a suicide risk by the investigator, currently (or within 30 days before the planned start of treatment) receiving an investigational drug, or currently using an experimental medical device.
Study Design
This randomized, double-blind, placebo-controlled crossover study in adult participants with ADHD was conducted to assess clinical efficacy, the time of onset, and time course of efficacy of PRC-063 over 16 hr, as measured during two AWE sessions by the PERMP total score (PERMP-T).
The study timetable, as shown in Figure 1, shows the screening, baseline, open-label, dose-optimization visits with weekly (3-10 days) ADHD assessments, and the double-blind crossover period with two AWE sessions.
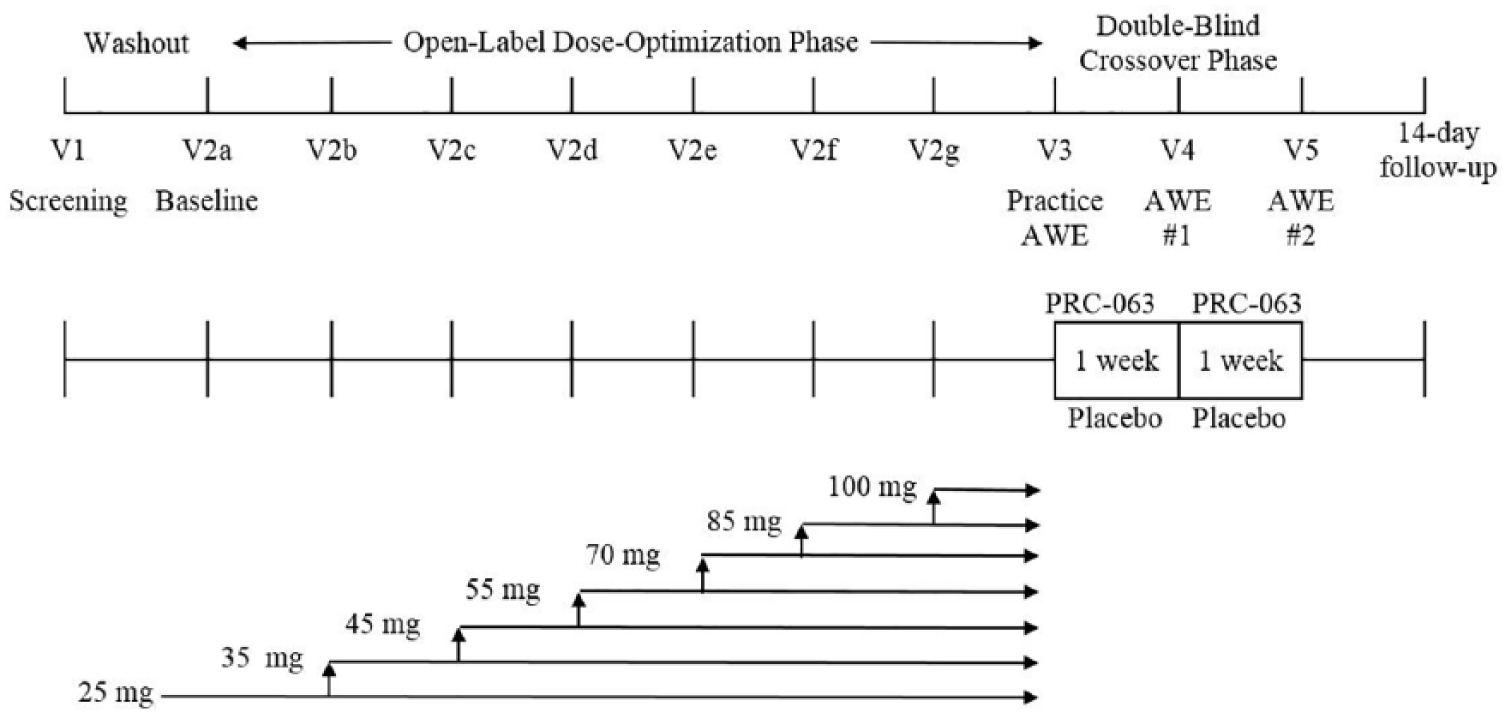
Study timetable.
Following screening and after a 7-day washout period, eligible participants entered the dose-optimization phase. Assessments during the dose-optimization phase included ADHD-5-RS, Clinical Global Impressions-Improvement Score (CGI-I), practice PERMP tests, Pittsburg Sleep Quality Index (PSQI), Columbia Suicide Severity Rating Scale (CSSRS) suicidality assessment (Posner et al., 2011), concomitant medication, and adverse events. An optimized dose was defined as a combination of clinical judgment and a ≥30% decrease from the participant’s baseline ADHD-5-RS score. Once participants reached their optimal dose, they were maintained on that dose for the remainder of the study.
Once the open-label, dose-optimization phase was completed, participants attended a half-day practice AWE session and entered the double-blind phase.
The double-blind phase consisted of either ACTIVE-to-PLACEBO (1 week of active treatment, one AWE session on active treatment, 1 week of placebo treatment, and one AWE session on placebo treatment) or PLACEBO-to-ACTIVE (1 week of placebo treatment, one AWE session on placebo treatment, 1 week of active treatment, and one AWE session on active treatment). The AWE sessions included objective, individualized math tests (PERMP), and concurrent behavioral ratings by blinded observers on the Swanson, Kotkin, Agler, M-Flynn, and Pelham Scale (SKAMP; Wigal et al., 1998) at specific time points throughout the 16-hr period (pre-dose and 1, 2, 5, 8, 11, 14, and 16 hr post-dose), designed to evaluate onset and duration of effect of PRC-063, and Time Segment Rating System (TSRS) assessments at 0.5, 4.0, 7.0, and 13.0 hr post-dose (intended to provoke ADHD-like behaviors by providing adults with opportunities during tasks to show symptoms and functional impairment due to ADHD). Once the double-blind phase was completed, there was a 14-day safety follow-up contact.
All available doses of PRC-063 were used in this crossover study (25 mg, 35 mg, 45 mg, 55 mg, 70 mg, 85 mg, and 100 mg per day) given orally once daily in the morning, no later than 10:00 a.m., and were provided in bottles of 10 capsules for each 1-week dispensing interval. This study included a 7-day double-blind, placebo-controlled washout period between the active AWE session and placebo AWE session for participants in the ACTIVE-to-PLACEBO sequence. Participants in the PLACEBO-to-ACTIVE sequence had a similar 7-day double-blind, stable dose of active medication between the placebo AWE session and active AWE session.
Outcome Measures
The primary outcome measure was the mean within-subject PERMP-T between treatments. The PERMP-T is the sum of the number of math problems attempted (PERMP-A) and the number of math problems answered correctly (PERMP-C) in a 10-min session.
Because adults may not have recent experience with performing math calculations by hand, at least 10 practice PERMP tests were conducted prior to the first full AWE session, including at least three tests during the half-day practice AWE session. Practice PERMP tests are identical in format to the PERMP used for each AWE session. It has been recommended that between six and 10 PERMP practice sessions are necessary to eliminate the practice effect (Wigal & Wigal, 2006).
The onset and time course of efficacy of PRC-063 compared with placebo was measured by comparing the PERMP at pre-dose and 1.0, 2.0, 5.0, 8.0, 11.0, 14.0, and 16.0 hr post-dose between each AWE session.
In addition, blinded observed rated each participant’s behavior and deportment using the SKAMP at the same time points as the PERMP. The SKAMP is a 13-item scale that assesses impairment related to inattention and behavior problems in a classroom environment (Wigal et al., 1998). Higher ratings reflect greater impairment.
Participants’ ADHD symptoms were assessed at every visit with the ADHD-5-RS. The ADHD-5-RS transforms the 18-item DSM-5 diagnostic criteria for ADHD, as licensed from the APA, to an 18-question scale (0 = not present; 3 = severe; total score of 0 to 54) used to assess the symptoms of ADHD and is similar to the ADHD-RS-IV (APA, 2013; DuPaul, Power, Anastopoulos, & Reid, 1998). During the open-label dose-optimization phase, ADHD-5-RS ratings were used to determine the titration of PRC-063. During the double-blind phase, while the participant was receiving the blinded medication, the ADHD-5-RS was used to assess participants’ ADHD behavior.
Following the washout period, clinicians rated the overall severity of ADHD using the CGI-Severity scale (1 = normal, not at all ill; 7 = severely ill). At each visit thereafter, clinicians rated improvement relative to baseline using the CGI-Improvement scale (1 = very much improved; 7 = very much worse).
During the double-blind AWE sessions, participants rated their ADHD behavior over the previous week using the Conners’ Adult ADHD Rating Scale-Self (CAARS-S). The CAARS-S consists of five subscales (Inattention/Memory Problems, Hyperactivity/Restlessness, Impulsivity/Emotional Lability, Problems With Self-Concept, and ADHD Index), which were converted to T scores for analysis. T scores are standard scores, z-shifted and scaled to have a mean of 50 and SD of 10.
Safety outcome measures included the CSSRS (a two-page clinician-administered rating scale to assess the presence of suicidal ideation or behavior), PSQI (Buysse, Reynolds, Monk, Berman, & Kupfer, 1989; a 10-question self-assessment of sleep habits and quality), concomitant medications, 12-lead ECGs (performed at screening and end of study), clinical chemistry, hematology, urinalysis, physical exam, vital signs, and spontaneously reported adverse events.
Participant sleep quality was measured using the PSQI at screening (to assess pre-study sleep quality over the past 6 months), after washout (to establish a baseline sleep quality when not receiving ADHD medication), and at end of study (to assess sleep quality on study medication). The global PSQI score was derived as the sum of the following seven component scores: overall sleep quality, sleep latency, duration of sleep, sleep efficiency, sleep disturbance, medication required to sleep, and dysfunction due to sleepiness during the day. The global score is on a scale from 0 (better) to 21 (worse), whereas all component scores are on a scale from 0 (better) to 3 (worse). Global PSQI scores greater than 5 are associated with poor sleep quality and Global PSQI scores less than or equal to 5 are associated with good sleep quality (Buysse et al., 1989).
Statistical Analysis
The safety population included all patients who took at least one dose of study medication. The full analysis (FA) population included all patients who had at least one PERMP rating for each visit of the double-blind phase.
Sample size calculations were based on detecting the mean difference between active and placebo for the primary efficacy endpoint of the PERMP-T. Assuming a treatment difference of 17 between PRC-063 and placebo and a SD of the treatment difference of 40, 46 completed participants were needed (23 in each of two treatment sequences) to maintain a two-sided family-wise Type I error rate of 0.05, with 80% power.
The primary efficacy analysis of PERMP-T was based on a repeated measures mixed-effects ANOVA model for the FA population. The model contained fixed effect terms for treatment, period, hour, treatment-by-time, and sequence. Two-sided 95% confidence intervals were calculated for the difference between treatments at each time point for the PERMP-T assessments. The “time of onset of efficacy” was defined as the earliest time point when the difference on the PERMP-T between an active dose group and placebo first became statistically significant. The “time course of efficacy” (i.e., duration of efficacy) was defined as the latest time point when the difference on the PERMP-T between an active dose group and placebo was statistically significant. These analyses were repeated for the PERMP-A (number of problems attempted) and PERMP-C (number of problems correct) as a secondary outcome measure.
Additional analyses using the mixed-effects model and analytical approach described above for the primary efficacy endpoint were also used to evaluate the SKAMP combined score, as well as the attention and deportment sub-scores.
Results
Participants
Of the 59 adults who were randomized, 53 completed the Titration Phase; five (8.5%) withdrew due to adverse events and one due to lack of response. Fifty-three adults entered the double-blind phase and were administered one of the seven strengths of PRC-063 or placebo. A further eight participants failed to complete the double-blind portion of the study; one missed the baseline time point on one AWE session, two withdrew due to adverse events, and five others failed to complete the study due to participant choice. Of the 45 participants who completed the study, two were on 45 mg, five were on 55 mg, 15 were on 70 mg, 11 were on 85 mg, and 12 were on 100 mg (see Table 1 for participant demographics). Most patients were female (64.4%). The average age of participants completing the study was 32.2 years, with 24 adults between 18 and 29 years, 18 between 30 and 49 years, and three between 50 and 59 years. The most common ADHD diagnosis subtype was combined type with 34 (75.6%), 11 (24.4%) were diagnosed as inattentive type, and none were hyperactive/impulsive type. The most common psychiatric comorbidities were “depression” (n = 5) and “anxiety” (n = 4). The mean score for CGI of Severity of ADHD at Baseline for all participants who completed was 4.5 ± 0.55, with 21 (46.7%) rated as “markedly ill,” 23 (51.1%) rated as “moderately ill,” and one (2.2%) rated as “severely ill.”
Participant Demographics (N = 45).
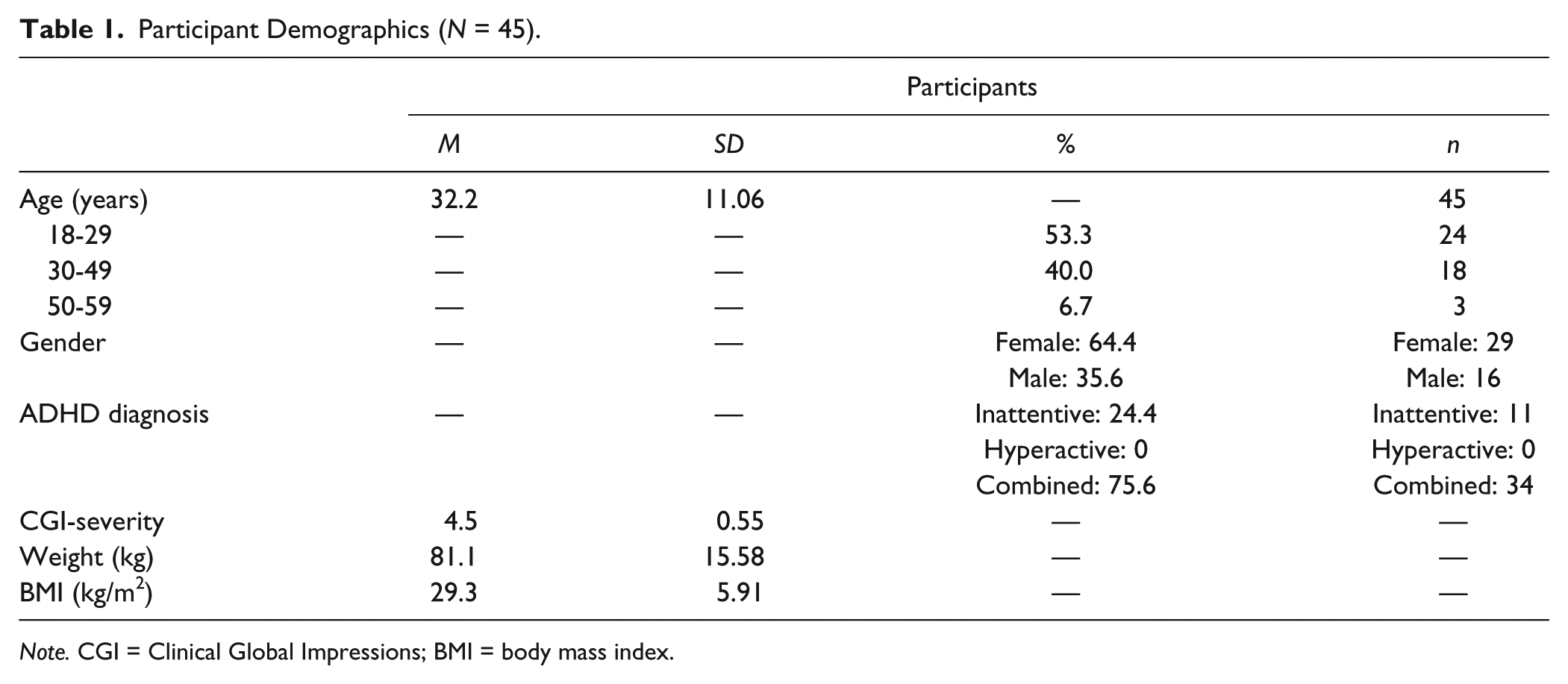
Note. CGI = Clinical Global Impressions; BMI = body mass index.
None of the participants received incorrectly assigned treatment and no deviations (e.g., missed visits) were considered sufficient to affect study efficacy or safety. All 59 participants who received any dose of PRC-063 were included in the safety population.
Efficacy
PERMP-T
When participants were treated with PRC-063, they had improved efficacy compared with placebo, as measured by the mean PERMP-T across all time points (268.7 ± 11.24 vs. 255.6 ± 10.87; p = .0064). There was no sequence effect (p = .1322), but there was a period effect observed (p < 0.0001).
Secondary analyses included the onset and time course of efficacy of PRC-063 compared with placebo as measured by the PERMP-T pre-dose and 1.0, 2.0, 5.0, 8.0, 11.0, 14.0, and 16.0 hr post-dose. Time of onset and the time course of efficacy (duration) were defined as the first and last time point, respectively, measured in the study where the difference in the change from pre-dose least square mean PERMP-T outcome between the active treatment and placebo was statistically significant. Onset of effect of PRC-063 occurred by 1.0 hr, the first time point measured in the study, and the between-group difference in PERMP-T remained significantly different at 2.0, 8.0, 11.0, and 16.0 hr post-dose, the last time point measured (Figure 2).
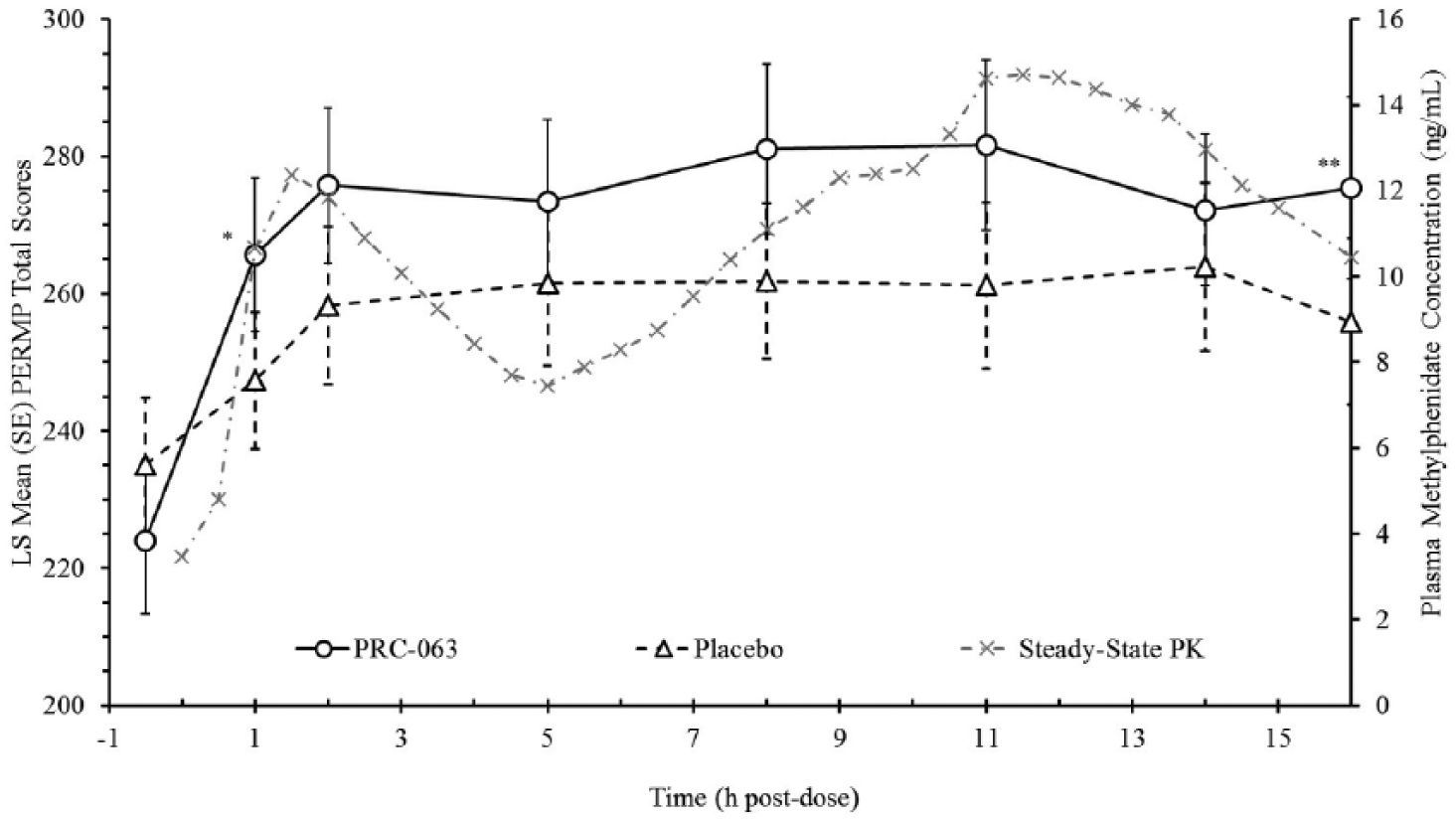
PERMP-T across AWE sessions.
SKAMP ratings
During each AWE session, SKAMP ratings were collected within each PERMP classroom session (at pre-dose, 1.0, 2.0, 5.0, 8.0, 11.0, 14.0, and 16.0 hr post-dose). Lower scores indicate improved attention and/or behavior. Figure 3 summarizes the Combined Score for the SKAMP for the FA population across all time points and for PERMP sessions. When SKAMP-C scores were analyzed across all PERMP time points with Hour 0 as baseline, PRC-063 had improved ratings of observed ADHD behavior compared with placebo (−3.5 ± 3.99 vs. 0.1 ± 4.41; p < .0001). There was no sequence effect (p = .6204) or period effect (p = .7468). SKAMP ratings were also conducted during TSRS tasks (at 0.5, 4.0, 7.0, and 13.0 hr post-dose). No significant differences on the SKAMP-C scale were observed at any of the TSRS time points (PRC-063: 15.2 ± 4.49; Placebo: 15.3 ± 5.05; p = .8010.
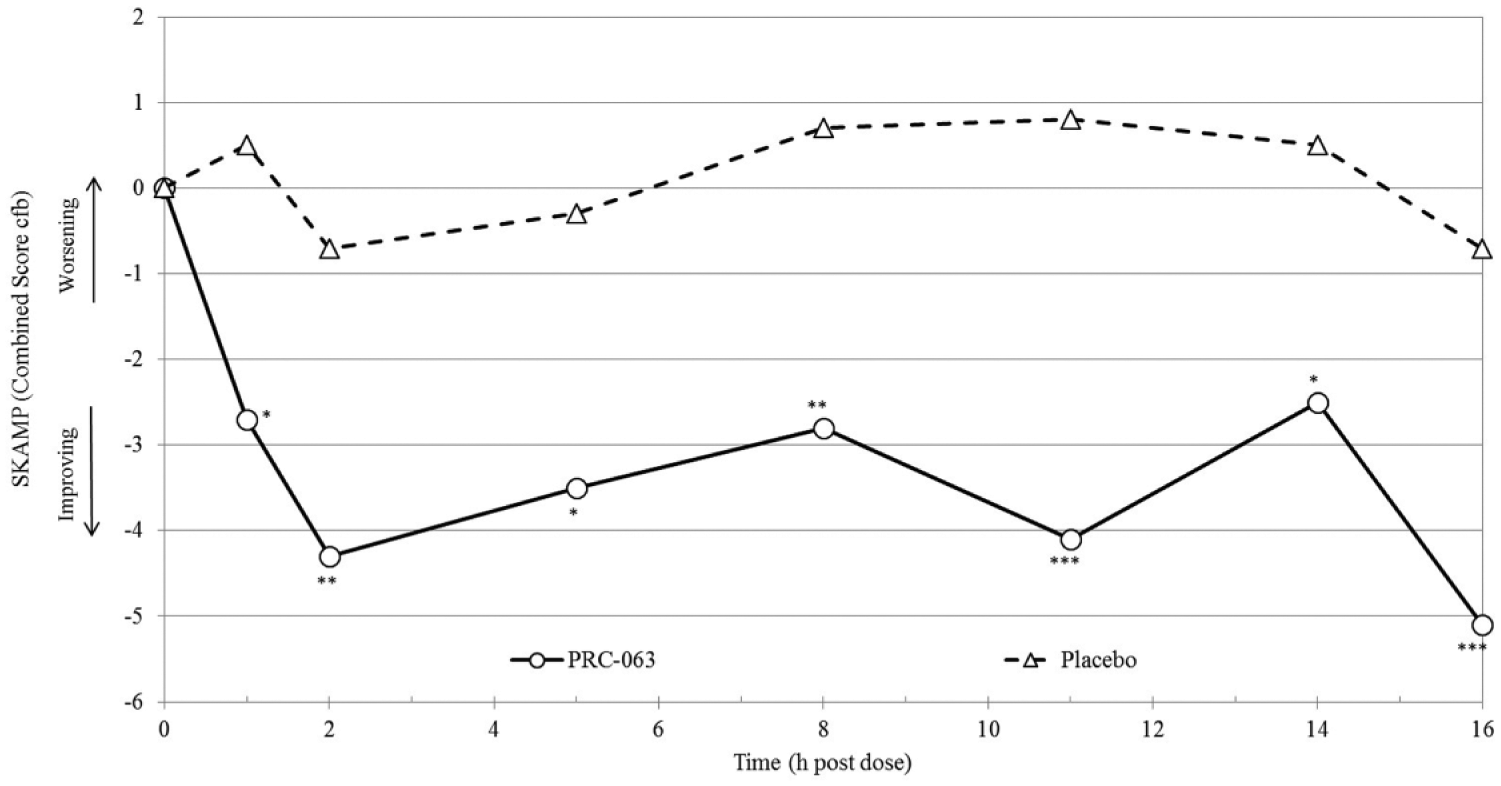
SKAMP-Combined Score across AWE sessions.
Symptom Rating Scales
ADHD-RS-5 scores
The total score and the subscale scores from the clinician-administered ADHD-5-RS were compared between treatments during the AWE randomized phase. When participants received PRC-063, they had fewer symptoms of ADHD compared with placebo during the blinded AWE phase of the study, as rated by clinicians using the ADHD-5-RS Total Score (16.8 ± 9.95 vs. 30.6 ± 11.76; p < .0001; Table 2).
Secondary Outcome Measures (N = 45).
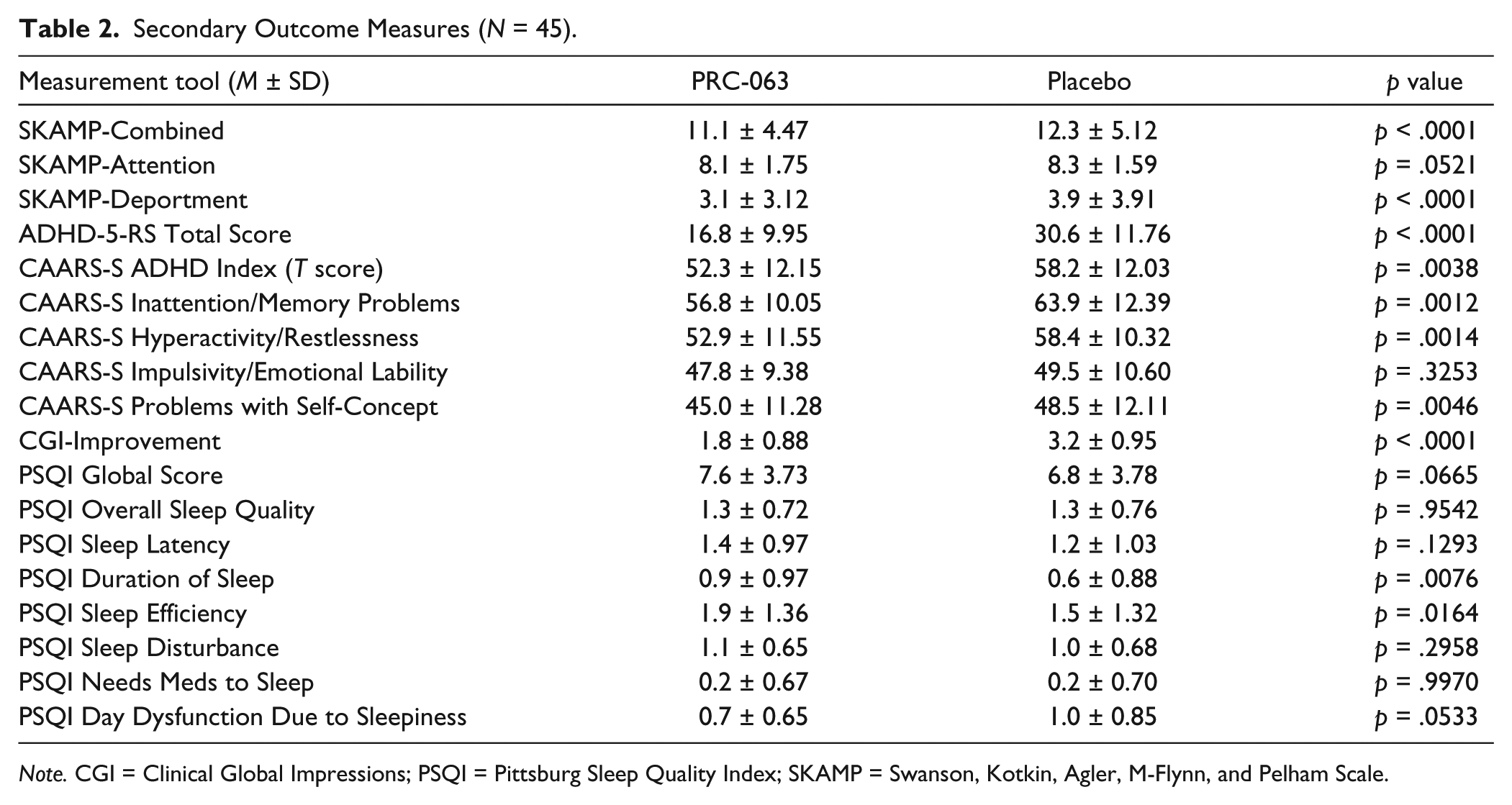
Note. CGI = Clinical Global Impressions; PSQI = Pittsburg Sleep Quality Index; SKAMP = Swanson, Kotkin, Agler, M-Flynn, and Pelham Scale.
CAARS-S
Participants rated their ADHD symptoms significantly improved using the CAARS-S when receiving PRC-063 compared with placebo during the blinded AWE sessions. Table 2 summarizes all subscales of the CAARS-S. When participants received PRC-063, they had significantly lower T scores compared with when they received placebo as measured using the CAARS-S ADHD Index, mean difference (SD): −6.0 (1.96), p = .0038; Inattention/Memory Problems, mean difference (SD): −7.1 (2.05), p = .0012; Hyperactivity/Restlessness, mean difference (SD): −5.5 (1.62), p = .0014; and Problems With Self-Concept, mean difference (SD): −3.5 (1.17), p = .0046. There was no significant difference between treatments on Impulsivity/Emotional Lability subscale, mean difference (SD): −1.7 (1.75), p = .3253.
CGI-I
Table 2 summarizes the results of the CGI-Improvement scale during the randomized blinded AWE phase by treatment. A lower score indicates greater improvement. Participants receiving PRC-063 had a greater improvement in ADHD symptoms compared with placebo (1.8 ± 0.88 vs. 3.2 ± 0.95; p < .0001). In addition, 84.1% of participants on PRC-063 were rated as “much improved” or “very much improved” versus 22.3% of placebo participants (p = .0001).
Safety
PSQI
Table 2 summarizes the results of the PSQI during the randomized blinded AWE phase by treatment. A repeated measures mixed-effects model was used to compare continuous results between treatments. No difference in PSQI global score (p = .0665) or overall sleep quality (p = .9542) was observed between PRC-063 and placebo treatment. Mean duration of sleep was shorter when participants received PRC-063 versus placebo (p = .0076), although the mean score for both treatments was between the categories of greater than 7 hr of sleep per night and 7 hr of sleep per night. The mean ratio of hours in bed to hours asleep (sleep efficiency) was worse for participants receiving PRC-063 versus placebo (p = .0164), although the mean score for both translates to between 65% and 75% sleep efficiency. No differences between PRC-063 and placebo were observed for sleep latency, sleep disturbance, needs medications to sleep, or day dysfunction due to sleepiness.
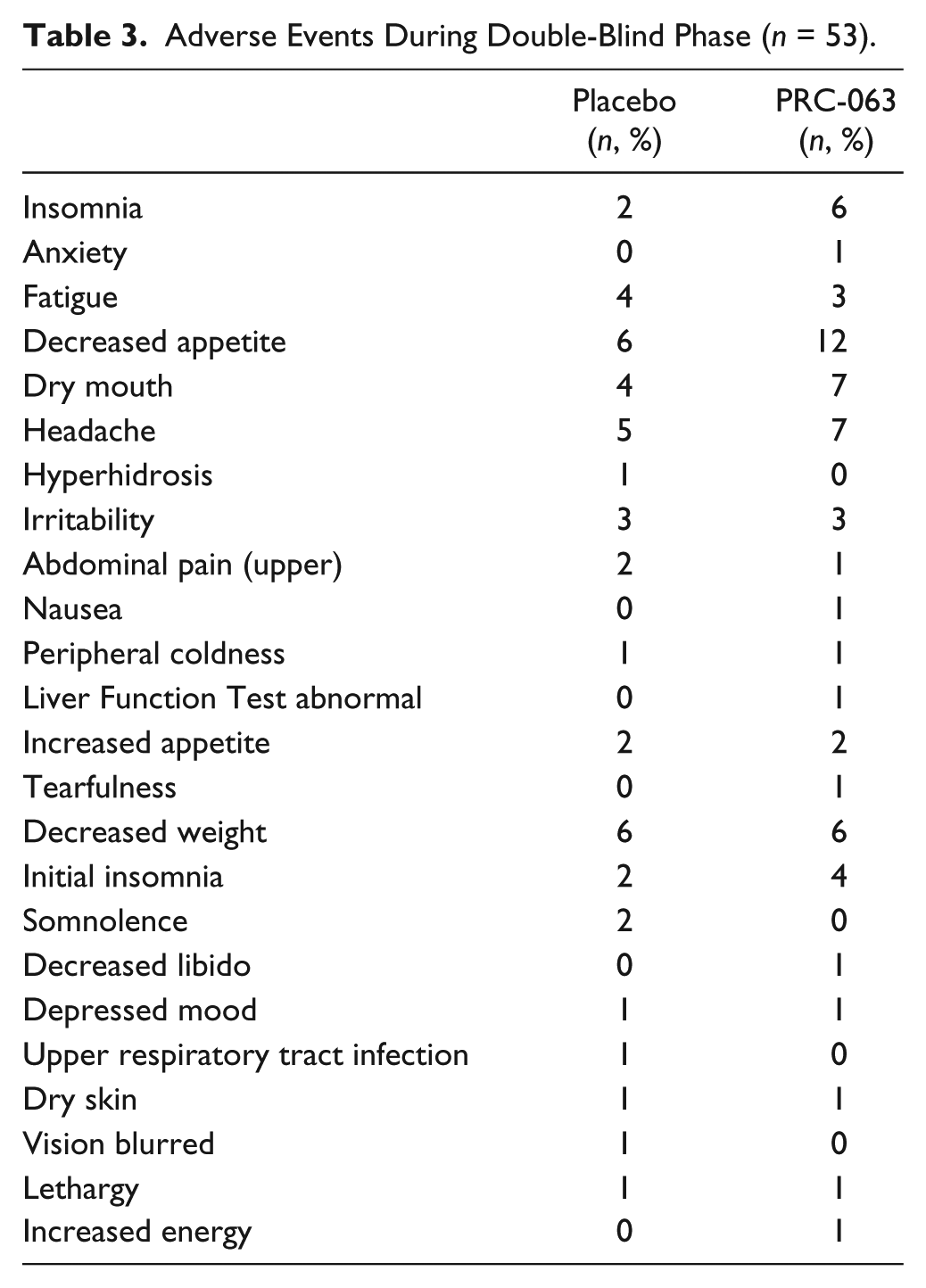
Eleven treatment-emergent adverse events (TEAEs) occurred in 10% or greater of participants receiving PRC-063 (n = 59): decreased appetite (47.5%), headache (39.0%), insomnia (30.5%), initial insomnia (28.8%), dry mouth (28.8%), upper respiratory tract infection (22.0%), nausea (18.6%), middle insomnia (16.9%), fatigue (16.9%), irritability (11.9%), and decreased weight (10.2%). Headache was the most frequent TEAE for placebo treatments, occurring in 8.0%. Adverse events that resulted in discontinuation were nausea (n = 1), bronchitis (1), gastroenteritis viral (1), viral infection (1), increased blood pressure (1), and hypomania (1), but none were classified as serious adverse events. TEAEs were experienced by 59.3% of participants when receiving 25 mg, 55.9% receiving 35 mg, 45.8% receiving 45 mg, 43.6% receiving 55 mg, 73.5% receiving 70 mg, 51.6% receiving 85 mg, 68.8% receiving 100 mg, and 40.0% of participants when receiving placebo. TEAEs were coded using the MedDRA dictionary Version 17.0. The percentage of participants experiencing severe TEAEs was 2.0% for the placebo group and 0.0% for the PRC-063-treated group.
Table 3 lists all TEAEs for each treatment group for the participants who participated in the double-blind phase (n = 53). To calculate these numbers, TEAEs that continued uninterrupted from the open-label, dose-optimization phase to the double-blind, crossover phase without a change in severity were counted only in the dose-optimization phase category. TEAEs with a change in severity across phases, or that resolved prior to the double-blind phase and then restarted in the double-blind phase, were counted in both phases. The most frequent TEAEs for participants during the double-blind, crossover phase (n = 53) were decreased appetite (n = 12, 22.6%), dry mouth (n = 7, 13.2%), insomnia (n = 6, 11.3%), headache (n = 7, 13.2%), and decreased weight (n = 6, 11.3%; Table 3).
Adverse Events During Double-Blind Phase (n = 53).
Participant Satisfaction
The following percentages relate to participant ratings of satisfaction of PRC-063 as “somewhat satisfied,” “satisfied,” or “very satisfied” on the Patient Satisfaction Survey: 100.0% for onset of action, 97.8% for duration of action, 100.0% for level of awareness, 73.3% for ability to fall asleep, 88.9% for level of lunch appetite, 86.7% for level of dinner appetite, 91.0% for overall side effects, and 88.9% for overall efficacy. Participants rated greater satisfaction with “how long the study medication lasted each day” on PRC-063 than on pre-study ADHD medication, with 88.9% of PRC-063 participants reported as “somewhat satisfied,” “satisfied,” or “very satisfied.” Scores did not significantly differ between pre-treatment ADHD medication and PRC-063 on any other PSS subscale.
Discussion
In this study, PRC-063 produced significantly greater PERMP-T improvement across the day than placebo in adults with ADHD. The superiority of PRC-063 was observed at 1 hr post-dose and continued through the 16 hr post-dose duration of the study. The effects of PRC-063 versus placebo on the SKAMP outcomes measured during the classroom sessions were consistent with the primary efficacy findings. Superiority for PRC-063 was observed at every time point measured throughout the day. This is the first study in adults with ADHD to demonstrate meaningful use of SKAMP measurements in the classroom component of the AWE study day (Biederman et al., 2005). These results provide additional detail regarding behavioral and attentional deficits observed in participants with ADHD across the life span. The findings suggest that the controlled, standardized laboratory setting adapted from use in children and adolescents with ADHD (Wigal & Wigal, 2006) provides robust measurement of adults with ADHD. This may be because, by design, the AWE provokes the display of impaired adaptation to environmental demands.
In addition to the improvements observed during the AWE sessions, participants exhibited significantly fewer ADHD symptoms in the week before the AWE while receiving PRC-063 than when they received placebo, as rated by blinded clinicians on the ADHD-5-RS. Participant self-rating measures on the AWE day (i.e., CAARS-Self) indicated fewer symptoms with PRC-063 versus placebo. This suggests that participants were aware that ADHD symptoms were improved throughout the AWE day. This finding is especially notable because adults with ADHD often struggle with self-awareness, particularly in the way their behavior may be affecting others around them.
Treatment with PRC-063 was generally well tolerated and similar to other long-acting stimulants (McCracken et al., 2003; Swanson et al., 2004; Wigal et al., 2010; Wilens et al., 2008). Rates of adverse events were much lower in the blinded assessment period than in the open-label titration period. This may have been due to participants having longer exposure to treatment by the time they reach the double-blind assessment period or because adverse events reduced in frequency as each participant’s dose was optimized. Consistent with previous reports of stimulant-based medications for ADHD, the most frequent treatment-emergent side effects observed during the blinded assessments included decreased appetite and weight, headache, insomnia, and dry mouth. None of these resulted in discontinuation. Although duration of sleep was shorter on PRC-063 than on placebo according to the PSQI, the mean duration of sleep on both treatments was between 6 and 7 hr per night, suggesting the difference was not clinically meaningful (Buysse et al., 1989). Likewise, while sleep efficiency (the ratio of hours of sleep to hours spent in bed) was lower for participants when on PRC-063 than when on placebo, the mean ratio was within the 65% to 75% range for both treatments. The lack of clinical significance of these sleep findings was confirmed by similar outcomes for the PSQI global score and overall sleep quality score for both treatments, suggesting that participants in both treatment groups can be characterized as “poor sleepers” as indicated by a global score greater than 5 (Buysse et al., 1989). This profile is consistent with previous reports in adults with ADHD (Philipsen et al., 2005; Mahajan, Hong, Wigal, & Gehricke, 2009; Sobanski, Schredl, Kettler, & Alm, 2007).
Limitations
Our conclusions are mitigated by several methodological limitations. First, based on the literature, we anticipated a 1:1 ratio of males to females but there was a slight overrepresentation of females in the study (64.4%). Second, although sufficient power was achieved to detect statistical significance in clinically relevant measures, negative findings should be viewed with caution. Third, a period effect (p = .0472) was observed for the PERMP-T, suggesting a practice effect may have occurred, despite the completion of a large number of practice tests during the dose-optimization phase of the study. It is unclear how many practice tests must be completed to eradicate this nuisance variable. However, no sequence effect was observed and the treatment effect was clearly discernible. In addition, measurements of aspects of attention and behavior that are independent of skill development, such as the SKAMP, were not subject to a period influence and supported the efficacy effect demonstrated by the improvement in PERMP-T. Finally, our study was limited to adults 18 to 60 years of age. Although we mention implications of these findings across the life span, other age groups were not a part of this study.
Conclusion
This study provides evidence for the safety and efficacy of a novel extended release methylphenidate treatment for adults with ADHD. PRC-063 has the potential of providing efficacy during early morning routines through to late evening, with improvements in the ability to initiate and stay on task and improvements in ADHD symptom control that occurred within 1 hr of dosing and continued throughout the day for up to and including 16 hr. This study also demonstrates the usefulness of the SKAMP in studies of time course effects of treatment in adults with ADHD. The safety profile of PRC-063 was generally safe and well tolerated, with fewer adverse events observed as the daily dose was optimized.
Footnotes
Acknowledgements
The authors acknowledge the contributions of Audrey Kapelinski (Licensed Clinical Social Worker), Nicole Schlaff, and all the investigators and patients.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Sharon B. Wigal has been an advisory board and speakers bureau member/consultant for Arbor, Attentiv, Eli Lilly, Ironshore, Neos, Neurovance, NextWave, Noven, NuTec, Pfizer, Purdue, Rho, Rhodes Pharmaceuticals L.P., Shionogi, Shire, Tris, and Vernalis, and has received grant and research support from Eli Lilly, Forest, Ironshore, the National Institutes of Health, Neurovance, NextWave, Noven, NuTec, Purdue Pharma, Rho, Rhodes Pharmaceuticals L.P., Shire, Sunovion, and Tris. Ann Childress has been an advisory board and speakers bureau member/consultant for Shire Pharmaceuticals, Pfizer, NextWave Pharmaceuticals, Sunovion, Ironshore, Rhodes Pharmaceuticals, Neurovance, Neos, Arbor, Tris, and Purdue Pharma and has received grant and research support from Shire Pharmaceuticals, Pfizer, Noven, NextWave Pharmaceuticals, Lilly USA, Forest Research Institute, Otsuka, Sunovion, Ironshore, Rhodes Pharmaceuticals, Theravance, Neurovance, Neos, Arbor, Tris, Purdue Pharma, Lundbeck, Alcobra, Medgenics, and Pearson. Graeme A. E. Donnelly and Joseph L. Reiz are employees of Purdue Pharma Canada.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by Purdue Pharma, Canada.