
Abstract
Previously, many studies have reported changes in the gut microbiota of patients with colorectal cancer (CRC). While CRC is a well-described disease, the relationship between its development and features of the intestinal microbiome is still being understood. Evidence linking Fusobacterium nucleatum enrichment in colorectal tumor tissue has prompted the elucidation of various molecular mechanisms and tumor-promoting attributes. In this review we highlight various aspects of our understanding of the relationship between the development of CRC and the alteration of intestinal microbiome, focusing specifically on the role of F. nucleatum. As the amount of F. nucleatum DNA in CRC tissue is associated with shorter survival, it may potentially serve as a prognostic biomarker, and most importantly may open the door for a role in CRC treatment.
Colon dysbiosis and colon cancer
The human gut is a habitat for bacteria, archaea, viruses, and fungi. Major colonization of the microbial communities in the human gut starts at birth and plays an essential role in maintaining gut homeostasis while constantly influencing human health, including metabolism, immunity, and inflammation.1–3 The human gut microbiome profile varies from person to person as environmental or lifestyle factors such as diet, smoking, and exercise can shape the diversity and the composition of the gut microbiome. The distinct human gut microbiome and their dysbiosis might have an impact on an individual’s health status by contributing to inflammation, oxidative stress, DNA damage, and immune invasion in the colorectal mucosa that requires deeper understanding.
Colorectal cancer (CRC) is the third most commonly diagnosed malignancy and the second most prevalent cause of cancer-related deaths globally. 4 Regardless of its complex etiology, the dysbiosis of intestinal bacteria plays a significant role in the initiation and development of CRC.
The causal relationship between gut microbiota dysbiosis and CRC has been demonstrated using mice model.5,6 Colonization and dysbiosis of the human microbiome in CRC indicate their potential role in CRC initiation, development, progression, and treatment outcome. 7 Previous work from our group identified a colonic mucosal dysbiosis in the aberrant crypt foci, the earliest neoplasia that can potentially be developed into CRC. 8 It has been shown that certain colon bacterial species such as Bacteroides fragilis, pks+Escherichia coli, Streptococcus gallolyticus, and Morganella morganii were abundantly present in the tumorous CRC lesions. 9
One of the most remarkable findings of CRC dysbiosis is the presence and enrichment of oral pathogens such as Fusobacterium nucleatum, Parvimonas micra, Peptostreptococcus stomatis, Peptostreptococcus anaerobius, Porphyromonas asaccharolytica, Solobacterium moorei, and Prevotella intermedia. 9 Particularly, the presence of F. nucleatum, has been strongly linked with CRC using both stool and colonic mucosal samples. 10 F. nucleatum promotes colon cancer progression by altering the colon mucosal microbiota through the enrichment of pathogens related to the development of CRC. 11 Most recently, the evidence showed that a clade of specific F. nucleatum subspecies was enriched in the CRC. 12
F. nucleatum, an oral anaerobic opportunistic microorganism, is enriched in both stools and tumor tissues of patients with CRC. 13 F. nucleatum is known as a core member of oral species both in oral health and periodontitis. 14 In the oral cavity, oral biofilm development occurs in a sequential and orderly manner. Typically, Streptococcus and Actinomyces arrive first on the enamel of teeth covered with pellicles that are derived from saliva. F. nucleatum acts as a bridging microorganism by providing receptors for so-called late colonizers of oral biofilm to adhere to the dental plaque which facilitates a microbiome shift from aerobic Gram-positive species abundant early colonizer community to facultative anaerobes and strictly anaerobic Gram-negative late colonizer such as Porphyromonas gingivalis, Tanerella forsythia, and Aggregatibacter actinomycetemcomitans. In this way, the capacity of F. nucleatum to coaggregate with other bacterial species is one of its major pathogenic properties.
The two key virulence factors of F. nucleatum are fibroblast activation protein 2 (Fap2) 15 and Fusobacterium adhesin A (FadA). 16 Both proteins act as clumping factors and contribute to pathogenicity, however, each virulence factor contains distinct characteristics. The pangenomic analysis revealed that fadA gene was well conserved across all 4 F. nucleatum subspecies (Fn subspecies animalis, Fn subspecies nucleatum, Fn subspecies polymorphum, and Fn subspecies vincentii) while fap2 gene was absent in some subspecies of F. nucleatum. 12 FadA protein exists in two forms, the intact pre-FadA and the secreted mature FadA (mFadA). 17 The mFadA arranges amyloid-like fiber structure that shares biochemical properties of amyloid found in Alzheimer’s, Parkinson’s, and prion diseases. 18 Fap2 on the other hand, interfere with Natural Killer cell killing of tumors when F. nucleatum is bound, therefore tumor is protected.
In the oral cavity, periodontal pathogen P. gingivalis directly interacts with F. nucleatum and synergistically initiates oral tumorigenesis. 19 These oral pathobionts can translocate from the oral cavity to the colon as more than a liter of saliva enters the gastrointestinal tract on a daily basis in an average adult. 20 It is not surprising to speculate that a bacterial tumorigenesis property may spread from oral cavity to other parts of the gastrointestinal tract. In fact, it has been reported that F. nucleatum strains from tumor tissue and CRC patient’s saliva had a similarity of 40%. 21
The oral microbial biomarkers have been investigated using saliva from CRC patients previously,22,23 however the direct comparisons of both oral sample and gut tissue microbiome analysis have just emerged recently.23–25
Epidemiological evidence
Many bacterial species have been implicated in the development of various malignancies. 26 Characterization of the composition of the microbiota in CRC has shown an enrichment of Fusobacterium sequences in tumors compared to normal pairs.27,28 While the Bacteroidetes and Firmicutes phyla were depleted in tumors, Fusobacteria have also been visualized within colorectal tumors using fluorescence in situ hybridization. 27 In addition, individuals with a mutation in the BRAF v600E gene had significantly higher odds of F. nucleatum enrichment. 28
In CRC, F. nucleatum is increased in the intratumoral microbiome and has the capacity to persist in metastatic lesions. 29 Overgrowth of F. nucleatum in CRC is also associated with tumor size, survival time, 30 and recurrent CRC post-chemotherapy. 31 The abundance of F. nucleatum in feces is strongly associated with the presence of CRC, but not with the presence of adenomas. 32 A study from China demonstrated that the infection rate and abundance of F. nucleatum were higher in carcinoma tissues than in adjacent normal tissues from CRC patients. 33 F. nucleatum is associated with the occurrence and progression of colon carcinogenesis, as it is detected in higher rates in high-grade dysplasia lesions, compared to initial neoplastic transformation.34,35 Moreover, no associations have been observed with dietary habits, lifestyle, or other conditions of the colon such as ulcerative colitis, 35 although its presence can aggravate ulcerative colitis through promoting gut microbiota dysbiosis and dysmetabolism. 36 CRC patients with undetectable or low F. nucleatum abundance, are three times more likely to have rectal versus colon cancer. 37 Put together, this suggests that Fusobacterium may not be a causal factor, but rather grows in favorable conditions caused by the malignant tumor.
F. nucleatum densely colonizes the oral cavity and is associated with periodontitis. However, the relationship between F. nucleatum in CRC and the oral cavity is not well understood. Patients with CRC have identical strains of F. nucleatum in their CRC and oral cavity. 21 This suggests that F. nucleatum in CRC originates in the oral cavity.
Although many studies have reported changes in the gut microbiota in patients with CRC,27,38,39 it is uncertain if the cancer is the cause or consequence of the change in the microbiota. 40 Despite both possibilities exist, F. nucleatum may be a prognostic biomarker of CRC. 41 This is important as it opens the door for the potential use of antibiotics in the treatment or prevention of CRC.
Bacterial mechanisms of tumorigenesis
A neoplasm arises through the process of carcinogenesis when a typically functioning tissue or organ undergoes hyperplasia. In this state, the cells proliferate abnormally and in an uncontrolled manner causing the formation of an abnormal mass in the biological system. 42 Numerous reports in recent years have demonstrated the involvement of bacteria in tumor initiation, progression, metastasis, and resistance to treatment.43–46 The alterations in gut microbiome lead to various physiological changes in the body, such as metabolic changes, activation of immune cells, and generation of pro-inflammatory responses, which ultimately generate a microenvironment responsible for the tumor initiation.47,48 Various signaling pathways appear responsible for the development of CRC due to the infection of F. nucleatum.
Activation of oncogenic pathways by F. nucleatum
F. nucleatum promotes CRC cell adhesion to endothelial cells and facilitates extravasation and metastasis. 49 The pathogenic process of F. nucleatum begins with its adherence and invasion of the epithelium. 50 Its FadA adhesion protein plays a major role in the binding of the bacteria to the epithelial cells.16,51,52 This protein is present in two forms, the first form is pre-FadA, which attaches to the epithelial membrane while the second mature form of the protein is secreted. 17 Both forms of FadA protein combine and form an active complex known as FadAc, which is internalized. The binding of the FadA protein on endothelial cells occurs due to the cadherins, which are cell adhesion glycoproteins and bind to cytoplasmic proteins like β-catenin. 53 The invasion of FadA into the endothelial cells causes the stimulation of inflammatory signals and results in boosting the genes of inflammatory cytokines interleukin-8 (IL-8), interleukin-10 (IL-10), tumor necrosis factor-a (TNF-α), and nuclear factor kappa B (NF-κB).54–57 The complete process generates a microenvironment that ultimately becomes tumor-supportive and down-regulates the antitumor immune response leading to the initiation and progression of CRC Figure 1.
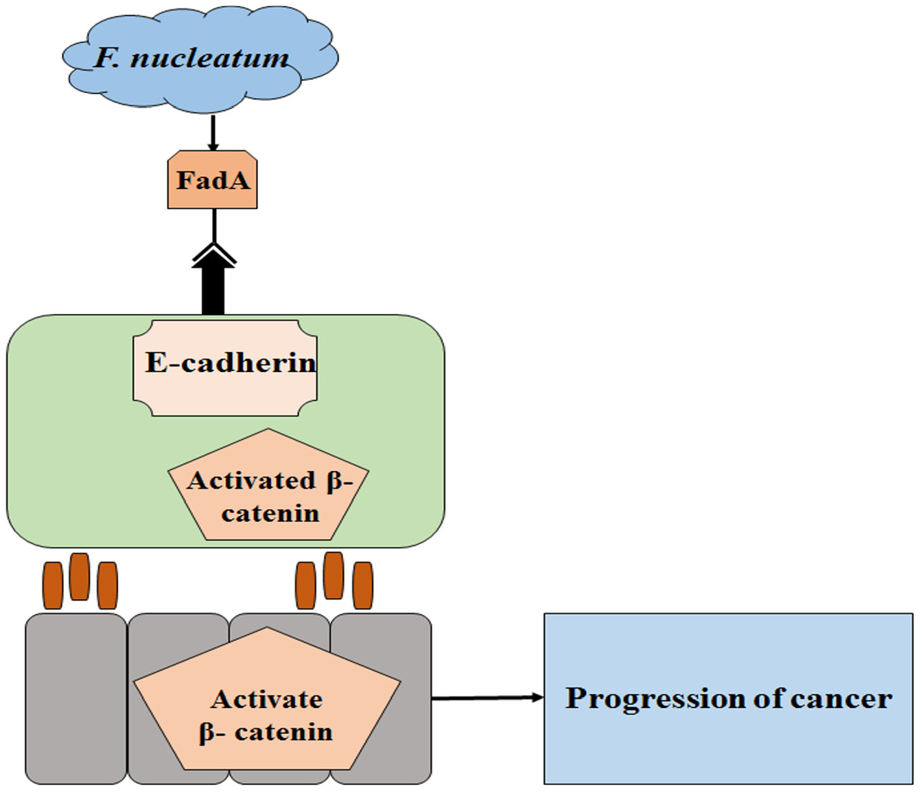
Potential mechanism of Fusobacterium nucleatum activity in CRC. FadA binding to E-cadherin increases β-catenin and upregulates annexin A1 that drives epithelial cell proliferation in the host leading to the progression of CRC.
Alternatively, F. nucleatum can enter into host mutated endothelial cells. The internalization of FadA activates β-catenin-regulated transcription which facilitates the translocation of β-catenin from the cytoplasm to the nucleus, resulting in the expression of genes of wnt signaling pathway (wnt7a, wnt7b, and wnt9a), Myc and cyclin D1 oncogenes, T-cell factors (TCF1, TCF3, and TCF4), and transcription factors such as lymphoid enhancer factor (LEF-1). It also leads to an increased expression of NF-κB, various pro-inflammatory genes along with oncogenes.57,58
CRC oncogenic mechanism of F. nucleatum through Fap2 lectin-mediated pathway
Different sugar epitopes are aberrantly expressed on the surface of cancerous cells as compared to normal cells. In CRC, there is the overexpression of sugar moieties of D-galactose-(1-3)-N-acetyl-D-galactosamine (Gal-GalNAc), and these carbohydrate epitopes are detected by fusobacterial Fap2 hemagglutinin which shows specificity toward Gal-GalNAc. 59 The Fap2 lectin plays a major role in not only the binding of the bacterium to the host cell but also in the invasion of Fusobacterium Figure 2.15,60–62 Various reports have corroborated the binding of Fap2 with Gal-GalNAc as Gal-GalNAc expression is reduced upon O-glycanase treatment, and its binding with lectin becomes weak. Mutant strains of Fusobacterium lacking Fap2 or inactivated Fap2 showed reduced binding with Gal-GalNAc in mice models. 59 In summary, both adhesion proteins (FadA and Fap2) play important roles in the binding of the F. nucleatum on the host cell surface, invasion inside the host cell, and triggering the CRC related oncogenic signaling pathways.17,19
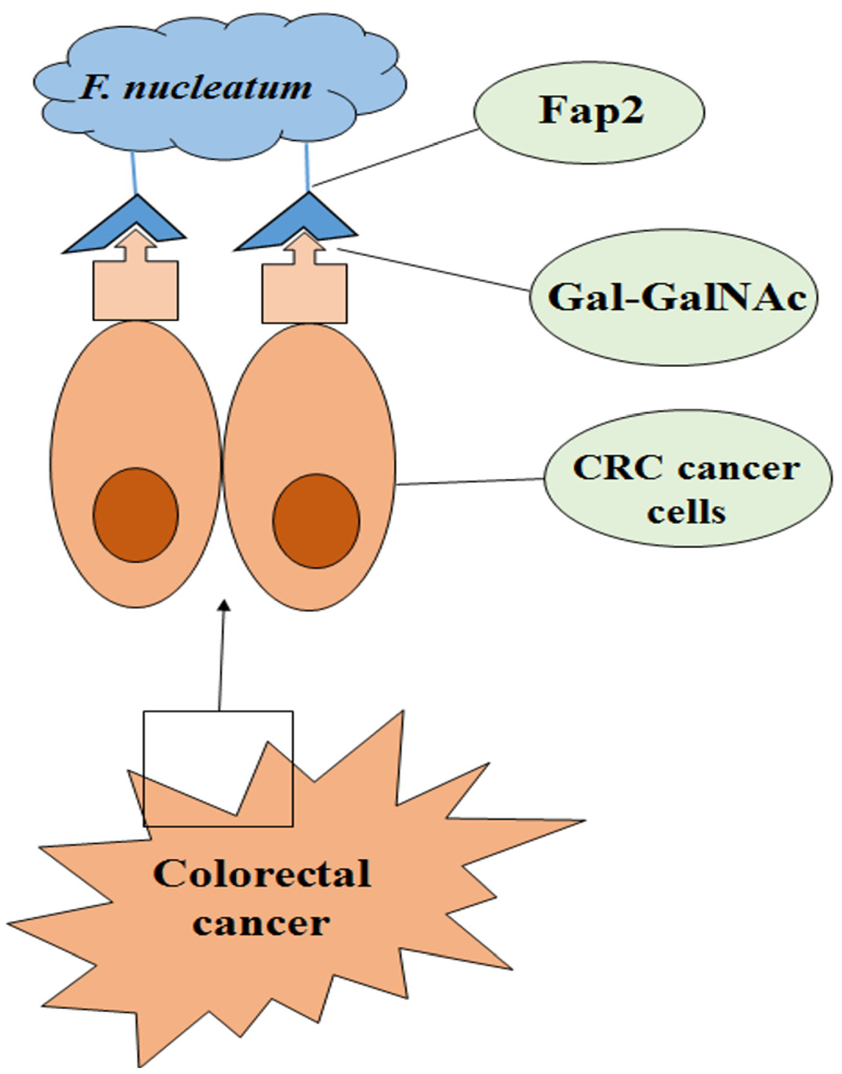
The aberrant expression of Gal-GalNAc on CRC cells provides the binding sites to Fusobacterium nucleatum. The CRC cells show the overexpression of Gal-GalNAc sugar moieties on cell surface. In F. nucleatum, Fap2 lectin binds specifically with Gal-GalNAc sugar epitopes which allows the bacterial cell binding and invasion to the CRC cells.
CRC oncogenic mechanism of F. nucleatum through activation of autophagy
In this pathway, F. nucleatum lipopolysaccharide (LPS) binds to Toll-like receptor 4 (TLR4). TLR4 is overexpressed in CRC cells and plays a major role in tumor initiation and development in the host cells. After binding of the Fusobacterium LPS to the host cell, microRNA-21 (miR-21, a chief promoter of colitis-associated colon cancer) expression increases, which can reduce the levels of RASA1 (responsible for the cell growth, proliferation, differentiation, and apoptosis) Figure 3a, ultimately affecting the MAPK pathway.43,63-65 The inhibition of miR-18* and miR-4802 expression by F. nucleatum via TLR4/MyD88 immunological signals induces CRC chemotherapy resistance by preventing apoptosis and promoting autophagy in the cancerous cells Figure 3b. 31
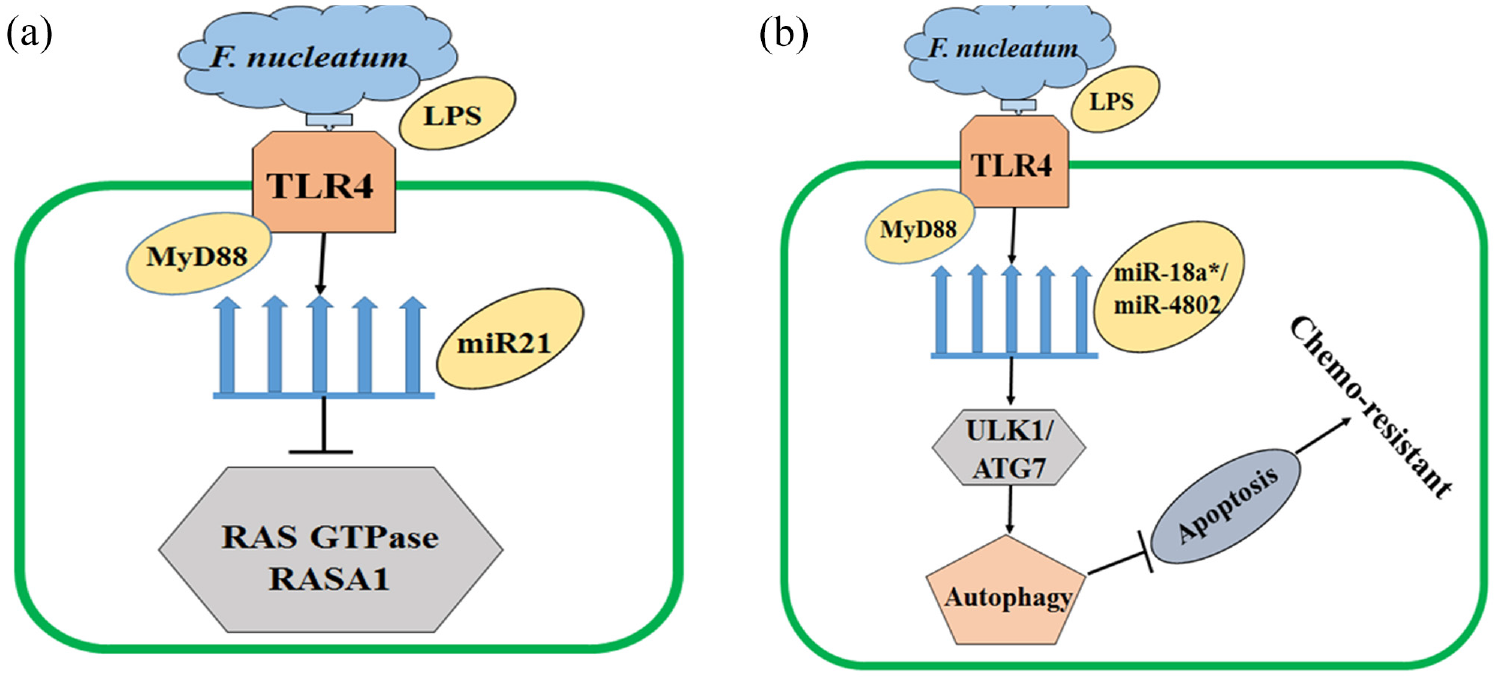
Fusobacterium nucleatum plays a vital role in regulation of CRC outcomes. (a) F. nucleatum can promote the proliferation of CRC cells by activating TLR4 and MYD88 signals, upregulating miR21 expression, and suppressing RAS GTPase RASA1 level. (b) F. nucleatum induces recurrence of CRC in the host cells after chemotherapy. The F. nucleatum activates TLR4 and MYD88 signals and reduces the expression of miR-8a* and miR-4802, respectively which activates autophagy, resists the apoptosis of cancerous cells, and induces chemo-resistance in the host cells.
F. nucleatum stimulates the growth of CRC cells without affecting the pre-cancerous adenoma cells. Annexin A1 is a key component for this stimulatory effect. Annexin A1 is specifically expressed in proliferating CRC cells and its expression is a predictor of poor prognosis. 57 The FadA adhesin from F. nucleatum upregulates Annexin A1 expression through E-cadherin. This mechanism indicates that microbes such as F. nucleatum can act as cancer facilitators. 57
Immune alterations in the host
Besides being involved in the occurrence and metastasis of CRC through virulence factors, oncogenic microRNAs, intestinal metabolites, DNA damage, and other mechanisms. F. nucleatum can also regulate the host’s immune response. 66 A major mechanism by which F. nucleatum contributes to the carcinogenesis of CRC is by inducing inflammation and suppressing host immunity. 67 The bacterial adhesion to the intestinal epithelium can cause the host to produce inflammatory factors and recruit inflammatory cells, creating an environment that favors tumor growth. It also induces immune suppression at the level of the intestinal mucosa which further contributes to the progression of CRC.
Monocyte chemoattractant protein 2 (CCL8/MCP-2), leukemia-inhibiting factor (LIF), and epithelial-derived neutrophil-activating protein (CXCL5/ENA-78) are elevated in the peritoneal fluid in anastomotic leakage after colorectal surgery. 68 CCL8 in tumor-associated macrophages accelerates the progression of CRC. 69
F. nucleatum induces CCL8 expression in macrophages via the TLR4/NF-κB signaling pathway, which is inhibited by iron deficiency. High iron is associated with a worse prognosis in patients with CRC with F. nucleatum. Furthermore, patients with CRC with elevated serum transferrin saturation showed iron deposition in macrophages in the tumor microenvironment. Mechanistically, iron attenuates the inhibitory phosphorylation of NF-κB p65, augmenting tumor-promoting chemokine production in macrophages. 70
F. nucleatum induces a tumor microenvironment that diminishes the adaptive immunity against CRC. This occurs in a T-cell dependent manner, where F. nucleatum infection leads to T-cell depletion and enrichment of exhausted CD8+ and FoxP3+ regulatory T cells in the tumor microenvironment. 71 Compared with F. nucleatum-negative cases, F. nucleatum-high cases are inversely associated with the density of CD3+ T cells. 72 This is associated with poorer disease-free survival and overall survival in stage III CRC patients. 71 At the same time, F. nucleatum induces greater cytokine responses in a human colorectal cell line, HCT116 compared to other species of the genus Fusobacterium. 19 F. nucleatum host-cell binding and invasion induces IL-8 and CXCL1 secretion that also drives CRC cell migration, increasing metastatic potential and cell seeding, poor prognosis. 73 F. nucleatum may produce cancer-associated metabolites with a direct interaction with the host immune system that would allow the bacterium to escape host humoral immune response by the host. 74
F. nucleatum subspecies do not form biofilms in vivo or associate with the mucosa in mice. 75 Tumor-isolated strains belong to F. nucleatum subspecies animalis, which has very recently been shown to be composed of two clades. Microbiome analysis of human tumor tissue from 116 patients with CRC demonstrating that it is clade C2 the one that dominates the tumor niche. 12
Clinical relevance and strategies aiming to revert poor prognosis
Multivariate analysis has shown that differentiation and F. nucleatum-positive CRC are associated. 34 The amount of F. nucleatum DNA in CRC tissue is associated with tumor metastasis and shorter postoperative survival, 33 and may potentially serve as a prognostic biomarker.66,76F. nucleatum promotes CRC cell adhesion to endothelial cells and facilitates extravasation and metastasis by inducing ALPK1/NF-κB/ICAM1 axis. 49
Studies suggest that the bacterium is also localized primarily within the metastatic cancer cells rather than in the stroma. 29 The DNA sequences of the Fusobacterium at the primary and metastatic sites were nearly identical within individual patients, suggesting that the bacteria may be traveling with cancer cells through the bloodstream to the sites of metastasis. This has implications for developing cancer prevention and treatment strategies through targeting the colon microbiota with antibiotics. In fact, antibiotic treatment is able to slow tumor growth in mice carrying xenografts of F. nucleatum-positive human CRC. 29 Mice treated with erythromycin, which does not kill Fusobacterium, showed no effect on the growth of Fusobacterium-positive tumors, but metronidazole reduced both the number of Fusobacterium in tumors and the rate of tumor cell proliferation and tumor growth. Curiously F. nucleatum is associated with improved therapeutic responses to PD-L1 blockade in patients with CRC, enhanced the antitumor effects of PD-L1 blockade on CRC in mice, and prolonged survival. 77 This is in great contrast with the detrimental effect that F. nucleatum has on the efficacy of αPD-L1 therapy in esophageal squamous cell carcinoma patients. 78 F. nucleatum stimulates cell proliferation and promotes PD-L1 expression in CRC. 79
Potential antibiotic approaches to control F. nucleatum overgrowth in patients with CRC need to be very specific, as broad-spectrum antibiotics could kill beneficial species of bacteria in the colon which can harm the patients by impairing their response to cancer therapy. Intratumoral F. nucleatum load has a poor prognostic effect on CRC by increasing nerve invasion, vascular tumor thrombus, and microsatellite mutation. 80 Sonodynamic therapy appears promising in improving cancer treatment with fewer toxic side by eliminating intratumoral F. nucleatum. 81 Another strategy involves using bacteriophages for elimination of tumor-colonized F. nucleatum, which is tumor targeting strategies using F. nucleatum and Fap2. 82 This latter uses weakened tumor-enhancing fusobacteria with FadA mutations, or the use of competing tumor-colonizing Salmonella and Listeria.
Conclusion
Given that CRC is the second most common cause of cancer-related death in men and women combined, with increasing mortality by 2030 83 hence, it is important to understand what factors contribute to the development of CRC. Dysbiosis of the gut microbiome has been found to play a key role in the development and progression of CRC. Various strains of bacteria have been linked to colorectal carcinogenesis. Of these, F. nucleatum has been established as a key member of the CRC-associated bacteria due to its multiple cancer-promoting mechanisms, such as activating oncogenic pathways, inducing a pro-inflammatory environment, and suppressing host immunity. However, only a small fraction of the complex interplay between the gut microbiome, F. nucleatum, and the development of CRC has been described.
Further research is needed to better understand the influence of geography, race, gender, and dietary habits on this association, as well as how it is affected by cancer treatment. It is possible that in the near future, implementation of this knowledge may elucidate new methods for CRC screening/risk stratification (adding molecular profiling of the gut microbiome to already available stool-based screening tests); treatment (altering F. nucleatum and using it as a Trojan horse to deliver drugs into a tumor - given the bacterium’s intimate relationship with CRC); and prevention (using narrow-spectrum antibiotics to eradicate culprit bacteria or immunization with an antigen derived from F. nucleatum). While some of these ideas may appear remote, ongoing efforts are needed to reduce the future impact imposed by CRC.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Sangeeta Tiwari acknowledges grant awards from National Institute of Health NIGMS (SC1GM140968) to support this work.