
Abstract
Iron is an essential element for the biological processes of living organisms, including the production of crucial oxygen-carrying proteins, formation of heme enzymes, and playing roles in electron transfer and oxidation–reduction reactions. It plays a significant role in various cardiovascular functions, including bioenergetics, electrical activity, and programmed cell death. Minor deficiencies of iron have been found to have negative impact on cardiovascular function in patients with heart failure (HF). The contractility of human cardiomyocytes is impaired by iron deficiency (ID), which results in reduced mitochondrial function and lower energy production, ultimately leading to cardiac function impairment, contributing to significant morbidity and mortality in patients with HF. This review discusses iron homeostasis within the human body, as well as ID pathophysiology and its role in HF. Focusing on therapeutic approaches including iron supplementation and/or repletion in patients with ID and HF, comparing results from recent clinical trials. Intravenous (IV) iron therapy has shown promising results in treating ID in HF patients. Large, randomized trials and meta-analysis, like Ferinject Assessment in patients with ID and chronic HF, AFFIRM-AHF, IRONMAN, and HEART-FID have demonstrated the efficacy of IV iron supplementation with IV ferric carboxymaltose or IV ferric derisomaltose in reducing hospitalizations and improving quality of life in patients with Heart Failure with reduced ejection fraction (HFrEF), New York Heart Association (NYHA) II–III. However, survival and mortality have demonstrated no improvement during acute exacerbations of HF or in outpatient management. The potential benefits of IV iron across the entire HF spectrum and its interaction with other HF therapies remain areas of interest for further research.
Iron is a common element that is essential for the biological processes of living organisms. In the human body, iron is found in complex forms bound to proteins like hemoglobin and myoglobin, and other enzymes containing iron. The body relies on iron for the production of crucial oxygen-carrying proteins, including hemoglobin and myoglobin. Iron is also important for the formation of heme enzymes and other enzymes containing iron, which play roles in electron transfer and oxidation–reduction reactions.
Iron plays a significant role in various cardiovascular functions, including bioenergetics, electrical activity, and programmed cell death. 1 In the early postnatal phase, the replication of cardiomyocytes in the mammalian myocardium becomes extremely limited. The number of cardiomyocytes in a mature mammalian heart remains relatively stable throughout life. Most fully developed cardiomyocytes are in a state of growth arrest in phases G0 or G1. Under prolonged stress, there is a restructuring at the ultrastructural level where the loss of cardiomyocytes exceeds their renewal, leading to a gradual onset of heart failure (HF).
Previously, it was thought that the population of cardiomyocytes remains constant, maintaining a one-to-one ratio with capillary microvessels that supply oxygen and nutrients. Recent studies, however, have revealed that mammalian cardiomyocytes still retain some capacity for division, and there are native cardiac stem and progenitor cells (CSCs/CPCs) within the heart. 2 However, the rate at which these cells turnover is undeniably limited. Mesenchymal stem cells (MSCs) have also been shown to undergo cardiac differentiation.2–4 Thus, iron is essential for MSCs/CSCs/CPCs as well as mature cardiac cells, but excess iron can be toxic.
Maintaining a balanced level of iron, known as iron homeostasis, is vital for overall health, especially for cardiovascular well-being. Therefore, the human cardiovascular system has developed complex mechanisms, including redundant iron intake proteins and regulatory processes, to ensure a stable iron level in the body. Maintaining proper iron levels is crucial for cardiovascular health. The heart is negatively affected by both insufficient and excessive amounts of iron, which is partly due to heightened levels of oxidative stress. 5
Even a minor deficiency of iron has been found to have negative impacts on cardiac function in HF patients, according to clinical trials. 6 The contractility of human cardiomyocytes is impaired by iron deficiency (ID), which results in reduced mitochondrial function and lower energy production, ultimately leading to cardiac function impairment. 7
However, excessive amounts of iron in the body can be detrimental to the heart’s proper functioning. This is evident in cases where individuals with β-thalassemia suffer from transfusional iron overload (IO) and those with hemochromatosis experience primary IO. The toxic effects of IO can potentially lead to impaired vascular and valvular calcification, worsened development of atherosclerosis, myocardial ischemia/reperfusion, HF, arrhythmias, and an overall decline in patient health.5,8,9
Iron metabolism in the heart
Iron is an essential micronutrient that plays a fundamental role as a cofactor for enzymes involved in the electron transport chain, oxygen binding, DNA synthesis, and redox reactions. 10 Its regulation in the body is tightly controlled to prevent overload which can lead to oxidative stress and damage. These functions are particularly critical in the heart, an organ with extraordinary energy demands and a perpetual need for oxygen.
One of the most critical roles of iron in the heart is its involvement in oxygen transport. Iron is a central component of hemoglobin, the oxygen-carrying protein found in red blood cells. Hemoglobin’s ability to bind and release oxygen depends on the iron atom at its core. When blood reaches the heart, oxygen is released from hemoglobin and taken up by cardiac myocytes. This oxygen is essential for contractility, allowing the heart to pump blood efficiently to meet the body’s demands.
Most of the energy production in cardiac muscle cells happens in the mitochondria.11,12 Iron is a crucial component in the electron transport chain, which is a series of protein complexes present in the inner mitochondrial membrane. These complexes help in the transfer of electrons, which in turn generates adenosine triphosphate (ATP), the cell’s primary source of energy. A well-functioning electron transport chain is essential for maintaining cardiac contractile function and overall energy balance. 13
Iron plays an important role in the antioxidant systems of the heart. Although it is crucial for redox reactions in the electron transport chain, it can also cause oxidative stress if it is not regulated properly.11–14 The heart relies on different antioxidant enzymes, such as superoxide dismutase, to reduce the harmful effects of reactive oxygen species (ROS) produced during energy generation.11–13 Therefore, maintaining a delicate balance of iron is crucial to prevent excessive ROS and oxidative damage to the heart tissues.
The body’s concentrations and iron homeostasis are regulated through various mechanisms that manage the control of intracellular iron metabolism, transfer, uptake, and export, as well as intracellular storage. 5 The heart has a unique iron homeostasis that deserves attention. Cardiac myocytes require a huge amount of energy for proper functioning, which is only possible with iron-containing mitochondrial enzymes. Hence, cardiac myocytes are vulnerable to ID. However, iron import proteins are not negatively regulated by systemic iron, making cardiomyocytes poorly protected from IO. The iron importing, sequestering, and exporting proteins, along with the iron regulatory proteins (IRPs), are responsible for regulating the iron level in cardiomyocytes. 15 All these proteins have been discovered in the cardiac muscle and are currently under study to understand their role in local iron homeostasis. 15
Iron import proteins
The heart has five crucial proteins that help in importing iron. Iron can enter the heart muscle cell through the conventional way by binding to the transferrin 1 receptor (TfR1). Newborn mice that did not have cardiac TfR1 died within 2 weeks of birth because of serious HF. ID in the heart caused by the knockout of TfR1 in mice led to problems in oxidative phosphorylation, metabolic alterations, necrosis, and damage to the structure of mitochondria.15,16
However, four other import pathways facilitate cardiac myocyte iron entry: T-type calcium channel (TTCC), divalent metal transporter 1 (DMT1), L-type calcium channel (LTCC), and Zrt-, Irt-like proteins (ZIP) 8 and 14. 15
During early embryonic development, TTCCs are expressed in the heart. However, shortly after birth, they disappear. In adults, TTCCs can be found only in cells of the conductive system, such as sinoatrial pacemaker cells and Purkinje fibers, where they play a crucial role in pacemaking activity. Under certain pathological conditions such as myocardial infarction, ventricular hypertrophy, or IO, TTCCs can reappear in the cardiac ventricles. Studies have also revealed that TTCCs act as the primary gateway for iron to enter cardiomyocytes during IO conditions. 15
DMT1 is another protein playing a critical role in iron uptake be cardiomyocytes. Cardiac DMT1 protein is increased in iron-deficient rats and decreased in IO rats. 15
The LTCC is highly prevalent in the heart and plays a crucial role in regulating iron levels. In cases of IO, the heart suffers from systolic and diastolic dysfunction, bradycardia, myocardial fibrosis, and IO cardiomyopathy as iron mainly enters the heart through calcium channels. Treatment with LTCC blockers such as amlodipine, verapamil, and nifedipine can reduce iron influx into cardiomyocytes, improve cardiac function, and normalize mitochondrial function in IO conditions. It is noteworthy that in IO, the gene expression of LTCC remains unaffected, indicating that LTCC is not modulated by iron levels. 15
The ZIP family of metal transporters are found in all eukaryotic organisms. They play a crucial role in regulating the transport of zinc into the cell cytoplasm. However, recent studies suggest that certain members of the ZIP family may also transport other metals such as manganese, cadmium, and iron. 15 Two members of the ZIP family, specifically, ZIP8 and ZIP14 are expressed in cardiomyocytes and may facilitate cellular iron uptake. 15
Cardiac iron sequestration proteins
Once inside the cell, iron becomes part of the poorly defined labile iron pool (LIP) in the cytosol. Iron within the LIP acts as an intermediary and can either be stored in ferritin (where it remains chemically inactive) or proceed through biosynthetic pathways to create heme and iron-sulfur clusters in the mitochondria or iron-dependent proteins in the cytosol. 17 Excess iron is expelled from the cell through the Fe2+ exporter, ferroportin 1 (FPN1).18,19 Under normal conditions, the level of labile iron is rigorously regulated to prevent the formation of ROS.
Intracellular ferritin (FT) is a ubiquitous protein serving as a buffer against both ID and excess. FT can store up to approximately 4500 iron atoms (with a typical value of around 1500 atoms) in a soluble, nontoxic form and aids in transport. 20 FT plays a crucial role in iron metabolism by sequestering iron during IO conditions, with antioxidant effects, and releasing it during ID. 21
Human FT comprises 24 subunits of 2 protein chains: Ferritin H (FTH) and Ferritin L (FTL). The ratio of FTH to FTL varies among different cell types and depends on tissue conditions. 22 FTH contributes to maintaining the LIP and shields the cell against oxidative stress resulting from elevated levels of available Fe2+ for the Fenton reaction. Conversely, FTL, as apoferritin, is also present in blood; serum FTL concentrations correlate with various pathological conditions. 23
The regulation of cellular iron homeostasis occurs posttranscriptionally through the interaction of IRP 1 and 2 with iron-responsive elements (IRE) on mRNA of specific genes. This interaction modulates the synthesis of key iron metabolism proteins involved in uptake, storage, and release. 24 While IRP1 serves a dual function as an IRE-binding protein and cytosolic aconitase (c-aconitase, CA), IRP2 remains stable in hypoxic and iron-deficient cells but undergoes degradation in normoxemic cells. 25 In conditions of low cellular iron concentration, IRPs stabilize the mRNA of TfR1 and DMT-1 to promote iron influx, while simultaneously inhibiting mRNA translation of FPN1 and FT to impede iron efflux and storage, respectively.26,27
In cardiac myocytes, FPN1 is regulated by hepcidin, produced both in the liver and locally within the heart. Hepcidin induces the degradation of FPN1, leading to reduced iron efflux from the cardiomyocyte. Cardiac hepcidin plays distinct autocrine roles in regulating iron within cardiomyocytes, differing from systemic iron regulation. Unlike systemic hepcidin, cardiac-hepcidin protein is upregulated instead of downregulated by hypoxia to preserve cellular iron. 28 This mechanism involves the posttranscriptional regulation of hepcidin peptide by the hypoxia-inducible factor (HIF) system, which upregulates furin, a HIF1α target gene. 29
The heart’s high susceptibility to IO is due to its possession of redundant iron-importing mechanisms while having only one iron-exporting protein, FPN. Iron homeostasis is intricately regulated within cardiomyocytes, whereas the transport and utilization of iron within the mitochondrial compartment, the metabolic engine of the heart, are controlled by specific proteins such as mitochondria-specific ferritin (FTMT), frataxin, and mitochondrial ATP-binding cassette subfamily B (ABCB) transporters.30,31
IO exerts significant deleterious impacts on cardiac myocytes, owing to the accumulation of excessive iron. 32 This overload precipitates numerous adverse effects on these cells. Primarily, it incites oxidative stress by generating (ROS) via Fenton and Haber–Weiss reactions, leading to cellular damage involving proteins, lipids, and DNA, potentially impeding cellular functionality, and contributing to myocardial injury.33,34
Furthermore, IO disrupts mitochondrial function within these cells. Mitochondria, pivotal in (ATP) generation essential for myocardial contraction, face disturbance in respiration due to elevated iron levels, causing energy depletion and undermining cardiac efficacy. 35
Extended IO may culminate in iron accumulation within cardiac myocytes, initiating iron-induced cardiomyopathy, characterized by myocardial fibrosis, hypertrophy, and compromised contractile function, eventually leading to HF. 32
Additionally, excessive intracellular iron can perturb cardiac conduction, potentially resulting in arrhythmias, impeding efficient cardiac output, and posing life-threatening risks. 36
Moreover, IO-triggered oxidative stress can elicit programmed cell death-apoptosis or uncontrolled cellular demise-necrosis in cardiac myocytes, exacerbating myocardial tissue degradation and impairing cardiac function. 37
Precise management of iron levels and intervention in IO conditions are crucial to prevent or alleviate these adverse repercussions on cardiac health. 38
ID and contractility of human cardiomyocytes
How ID impairs cardiac function on a cellular level in the human setting is less understood. Nevertheless, ID is common in patients with HF, and it is associated with poor cardiac function and higher mortality. A study by Hoes and colleagues has shed light on this mechanism. 39 They highlighted the profound impact of cellular ID on the functionality and health of human cardiomyocytes independent of its effects on hemoglobin, demonstrating that ID in human cardiomyocytes triggers a hypoxic response, leading to mitochondrial dysfunction, decreased ATP levels, and impaired contractility and relaxation. Upon restoring iron levels, these detrimental effects were reversed. To induce ID, an iron chelator, Deferoxamine (DFO), was employed. Although the model’s severity may surpass typical ID in patients, it revealed insights into cellular responses. 39
The study emphasized DFO’s effectiveness in chelating iron within cardiomyocytes, leading to cellular iron depletion. Iron-depleted cardiomyocytes exhibited decreased ATP levels, potentially suggesting mitochondrial dysfunction, with a metabolic shift to anaerobic glycolysis. Notably, these cells faced challenges in transporting and utilizing oxygen, contributing to mitochondrial dysfunction. Complexes I–III (iron-sulfur clusters) were notably affected by DFO treatment, while heme-based complexes IV and V remained unaltered, consistent with previous findings in HF patients. 39
Upon replenishing ferritin levels with transferrin-bound iron, the adverse effects of ID on cardiomyocytes’ iron metabolism, ATP production, and mitochondrial respiration were mostly restored, indicating high reversibility. Clinical trials have shown improvements in exercise capacity and symptoms following ID reversal, suggesting the potential for cardiomyocyte recovery.40–42 However, certain gene expression changes persisted postiron restitution, potentially due to various stress responses.
The study revealed that iron-depleted cardiomyocytes generated less force and exhibited compromised diastolic function, further highlighting the impact of intracellular iron levels on cardiomyocyte performance. Morphological examinations revealed mitochondrial abnormalities and endoplasmic reticulum (ER) stress, indicating a link between severe ID and ER stress-induced cellular responses. Disrupted lipid handling and homeostasis were also observed in iron-deficient cardiomyocytes. 39
Overall, this research underscores the significant repercussions of cellular ID on human cardiomyocytes, affecting mitochondrial function, ATP production, and contractility, all of which can be reversed by iron replacement. These findings shed light on potential mechanisms for enhancing cardiac function through ID treatment and caution against strategies altering the hepcidin/ferroportin axis, which may inadvertently reduce intracellular cardiac iron levels, posing challenges to cardiac health. 39
Chung and colleagues investigated the impact of ID on cardiac function using a mouse model of dietary ID. 43 They found that ID anemia leads to a contractile deficit in the heart, affecting various cardiac parameters. Initially, the mice exhibited markers of tissue hypoxia and iron depletion in the heart without significant changes in heart rate. However, over time, iron-deficient mice developed increased ventricular filling during diastole and incomplete emptying during systole, resulting in a moderate reduction in left and right ventricular ejection fraction (EF). These findings contradicted the expected outcome based on the Frank–Starling mechanism and suggested the onset of systolic dysfunction. 43
Furthermore, the study found that iron supplementation with intravenous (IV) ferric carboxymaltose (FCM) reversed anemia, restored normal EF, and normalized calcium transient (CaT) amplitudes. However, not all cardiac parameters returned to normal levels after iron supplementation, indicating that certain changes persisted despite addressing the anemia. Mechanistically, the remodeling of calcium signaling in iron-deficient hearts did not involve myocyte hypertrophy or changes in certain cellular factors such as L-type calcium current, diastolic sarcoplasmic reticulum calcium leak, or cytoplasmic calcium buffering. Instead, substantive remodeling occurred in two key proteins: the Ryanodine receptor 2 (RyR2) channels and Sarcoplasmic/Endoplasmic Reticulum Calcium ATPase (SERCA) pumps. 43
The reduction in RyR2 activity and SERCA activity explained alterations in calcium signaling. ID caused a decrease in RyR2 expression and reduced RyR2 activity, influencing the CaT rise-time and fractional sarcoplasmic reticulum release. Additionally, a decrease in SERCA activity was observed, indicated by a right-shift in its calcium-activation curve. These changes led to a stable alteration in CaT amplitude and contraction, contributing to the observed systolic dysfunction in iron-deficient mice. Lower calcium affinity was associated with decreased phosphorylation of phospholamban at Thr17, suggesting a metabolic mechanism underlying SERCA remodeling. The findings highlighted the complexity of calcium signaling remodeling in ID anemia and its impact on cardiac function, offering insights into potential therapeutic targets.
In essence, the study demonstrated that ID anemia induces detrimental changes in calcium handling proteins (RyR2 and SERCA) within the heart, resulting in altered calcium signaling and subsequent systolic dysfunction. While iron replacement reverses anemia and partly restores cardiac function, certain cardiac alterations persist, emphasizing the multifaceted nature of the condition’s impact on heart health. 43
Diagnosis and treatment of ID in HF
It has been well recognized that ID is commonly noted among HF patients. ID can be both absolute and functional. Absolute deficiency signifies depleted iron stores with otherwise intact iron metabolism, while functional deficiency refers to a dysregulation between iron demand and supply.
Although the gold standard for diagnosing ID is a bone marrow biopsy, its invasiveness prompts the use of alternative blood biomarkers. Serum ferritin, despite its correlation with ferritin expression in storage tissues, must be cautiously interpreted in chronic and inflammatory states. Transferrin saturation (TSAT), indicating the percentage of transferrin bound to iron, serves as a marker of available circulating iron for cells. In HF, absolute ID is diagnosed with a ferritin cutoff of <100 mg/L, while functional deficiency is identified with normal serum ferritin (100–299 mg/L) and low TSAT <20%. 44
Other markers such as soluble transferrin receptor (sTfR) and hepcidin levels are also considered but are usually employed in research settings due to their specific nuances and limitations. 45 Mean corpuscular volume and mean corpuscular Hb concentration are unreliable in diagnosing ID in HF patients, and serum iron levels can fluctuate due to diurnal variations. Re-evaluation of ID should be considered periodically and after each HF-related hospital admission.
With the well-established parameters of ID as defined above, it is estimated that between 40%–50% of congestive HF patients have ID and the incidence is even higher among females and among patients with acute HF. 46 ID in congestive HF patients is noted even in nonanemic HF patients.
The reasons for the high ID are multifactorial but include occult blood loss possibly from antiplatelet medications and anticoagulants, poor dietary iron intake, chronic low-grade inflammation resulting in hepcidin-induced iron sequestration, and kidney disease.
Multiple studies have been conducted over the years seeking to determine if iron repletion, either oral or IV, improved morbidity or mortality in iron-deficient HF patients.
Oral iron
Oral iron is the first-line treatment option in non-HF patients and is very effective, widely available, and of low cost. The well-known problems with poor adherence, gastrointestinal side effects, and low intestinal absorption are what lead to the subsequent administration of IV iron in oral nonresponders.
Nevertheless, in non-HF populations, IV and oral iron repletion results in similar improvement in ferritin levels, TSAT, and hemoglobin improvements. Unfortunately, the same has not been the case with oral iron repletion in HF iron-deficient patients. Trails exploring the use of oral iron supplementation have been few, with limited patients and outcomes.
One of the first randomized studies was the IRON-HF study. Suitable patients (Left Ventricular Ejection Fraction (LVEF) <40%, NYHA Class II–IV, hemoglobin 9–12 g/dL, TSAT <20%, and ferritin <500 μg/L) were randomized in a double-blind fashion to oral ferrous sulfate or IV therapy with iron sucrose combined with placebo. The oral treatment arm of the study encompassed only 7 patients, where 10 patients were assigned to IV iron supplementation. Such small numbers make any conclusions suspect but 3 months after treatment, IV unlike oral iron therapy led to higher peak oxygen consumption. Compared with the oral iron treatment group, IV supplementation resulted in numerically higher TSAT and ferritin increase. 47
A second randomized much larger study was the IRONOUT HF: Oral Iron Repletion Effects On Oxygen Uptake in HF. In this study, patients underwent treatment with oral iron polysaccharide, 150 mg twice daily over 16 weeks, in a double-blind, randomized, placebo-controlled fashion. The primary endpoint was a change in peak oxygen uptake from baseline to 16 weeks. Secondary endpoints included changes in 6 min walk distance, N-terminal pro-brain natriuretic peptide (NT-proBNP) levels, and quality of life (QoL) assessment. Oral iron therapy did not significantly improve any of the study endpoints and minimally influenced iron storage. Of note is that the oral iron regimen was a twice-per-day schedule and not the now more efficacious schedule of once-per-day or alternate-day schedules which have been proven more effective. 48
Thus, current recommendations, despite limited data, urge IV iron supplementation for ID patients with chronic HF. 49
IV iron therapy
Several large randomized prospective studies have been conducted on the use of IV iron in HF.
The FAIR-HF (Ferinject Assessment in patients with ID and chronic HF) was the first large study investigating the use of IV FCM in patients with chronic HF. NYHA Class II with LVEF ≤40% or NYHA Class III and LVEF ≤45 with ID (ferritin level <100 μg/L or between 100 and 299 μg/L, if the TSAT was <20%), with or without anemia, were randomized in a 2:1 fashion to receive IV FCM or placebo. Among the patients receiving FCM, 50% reported being much or moderately improved, as compared with 28% of patients receiving placebo, according to the Patient Global Assessment (odds ratio for improvement = 2.51; 95% confidence interval (CI) = 1.75–3.61). Among the patients assigned to FCM, 47% had a NYHA functional class I or II at week 24, as compared with 30% of patients assigned to placebo (odds ratio for improvement by one class = 2.40; 95% CI = 1.55–3.71). Results were similar in patients with anemia and those without anemia. Significant improvements were seen with FCM in the distance on the 6-min walk test and QoL assessments. The rates of death, adverse events, and serious adverse events were similar in the two study groups. There were limitations of the study in that most patients were white and male, most with ischemic cardiomyopathy, and short follow-up. However, the study did confirm what earlier smaller studies had found. 50
AFFIRM-AHF
The AFFIRM-AHF trial 51 a multicenter study of 1108 patients hospitalized for acute HF at 121 centers in Europe, Israel, Lebanon, South America, and Singapore. All had ID, which was defined as serum ferritin <100 ng/mL (or 100–299 ng/mL if TSAT <20%). The average age was 71 years, and the average EF was 33%.
Before discharge, patients were randomized to receive IV FCM or placebo. The first treatment was administered shortly before hospital discharge, with a second treatment given at week 6 to patients whose ID persisted. Doses of FCM were determined by the patient’s body weight and hemoglobin value, with the average dose being 1350 mg. Eighty percent of patients had resolution of their ID with one or two treatments, with the remaining patients receiving additional IV doses at weeks 12 and 24.
At a follow-up of 52 weeks, the incidence of CV death was not different between the treatment and placebo groups. For the combined primary endpoint of total hospitalizations and CV death, the total number of events was numerically lower in those treated with FCM compared with placebo (rate ratio (RR) = 0.79; 95% CI = 0.62–1.01), driven by a significant 26% reduction in the risk of HF hospitalizations.
The IRONMAN published recently demonstrated potential benefits of IV iron. 52 Unlike AFFIRM-AHF, which enrolled patients at the time of an acute HF hospitalization, IRONMAN enrolled outpatients with chronic HF and used a different iron formulation, ferric derisomaltose, which can be given as a rapid, high-dose infusion.
IRONMAN enrolled 1137 patients between August 2016 and October 2021, randomizing them to usual care or the ferric derisomaltose infusion, with redosing at week 4, month 4, and then at 4-month intervals thereafter if ferritin remained low. All patients at baseline had a left ventricular EF of 45% or less and TSAT less than 20% or serum ferritin less than 100 µg/L. 52
At a median of 2.7 years, 336 primary endpoints had occurred in the iron group and 411 in the controls (RR = 0.82; 95% CI = 0.66–1.02), narrowly missing statistical significance. HF hospitalizations, expressed as the number of events per 100 patient-year, trended higher in the usual-care arm (20.9 vs 16.7; p = 0.085), whereas CV deaths were similar (24% vs 21%; p = 0.23). 52
The results of IRONMAN were similar to those of AFFIRM-AHF and added considerably to the growing evidence showing that IV iron supplementation in patients who have ID, HF, and reduced EF (or mildly reduced EF) reduces hospital admissions for HF and improves QoL.
Very recently, the completed HEART-FID study results were presented at the European Society of Cardiology in 2023. The HEART-FID study, the largest investigation to date on IV iron replacementin HF, aimed to validate earlier indications of benefits from smaller studies. 53
The study involved 3065 patients from 281 centers across 14 countries who had HF with reduced EF, ID, moderate to severe HF symptoms, and were on optimized therapy for at least 2 weeks. They were randomly assigned to receive IV FCM or a placebo. 53
Patients received initial doses at 0 and 7 days, followed by doses every 6 months based on their iron levels and hemoglobin. The study’s primary endpoint was a combination of all-cause mortality, HF hospitalizations at 12 months, and changes in walking distance over 6 months. The secondary endpoint was the time taken for the first HF hospitalization or cardiovascular death during the follow-up period.
IV iron replacement showed modest benefits in recently hospitalized patients with HF and ID, but the study failed to meet the specified, more rigorous, definition of significance (p = 0.01) on the primary endpoint. The trial also showed no statistical difference in the main secondary endpoint.
Finally, to clarify and characterize the effects of IV iron (FCM) on hospitalizations and mortality, a recent meta-analysis 54 of the three large randomized controlled trials (CONFIRM-HF, AFFIRM-AHF, and HEART-FID) was completed with a total of 4501 patients included.
The co-primary efficacy endpoints were (i) a composite of total/recurrent cardiovascular hospitalizations and cardiovascular death and (ii) a composite of total HF hospitalizations and cardiovascular death, through 52 weeks. Key secondary endpoints included individual composite endpoint components. Event rates were analyzed using a negative binomial model. Treatment-emergent adverse events were also examined.
FCM was associated with a significantly reduced risk of co-primary endpoint 1 (RR = 0.86; 95% CI = 0.75–0.98; p = 0.029; Cochran Q = 0.008), with a trend toward a reduction of co-primary endpoint 2 (RR = 0.87; 95% CI = 0.75–1.01; p = 0.076; Cochran Q = 0.024). Treatment effects appeared to result from reduced hospitalization rates, not improved survival. The treatment appeared to have a good safety profile and was well tolerated. Thus, in ID patients with HF with reduced left ventricular EF, IV FCM was associated with a significantly reduced risk of hospital admissions for HF and cardiovascular causes, with no apparent effect on mortality.
Few, if any studies, have addressed if IV iron replacement benefits also extend to patients across the entire HF spectrum to include those with HF with preserved EF or asymptomatic left ventricular dysfunction. Also, it is unclear whether IV iron is of benefit if patients are optimized on guideline-directed medical therapies, including SGLT2 inhibitors.
Table 1 outlines the key differences between the discussed trials.
Summary of key differences between discussed clinical trials and meta-analysis.
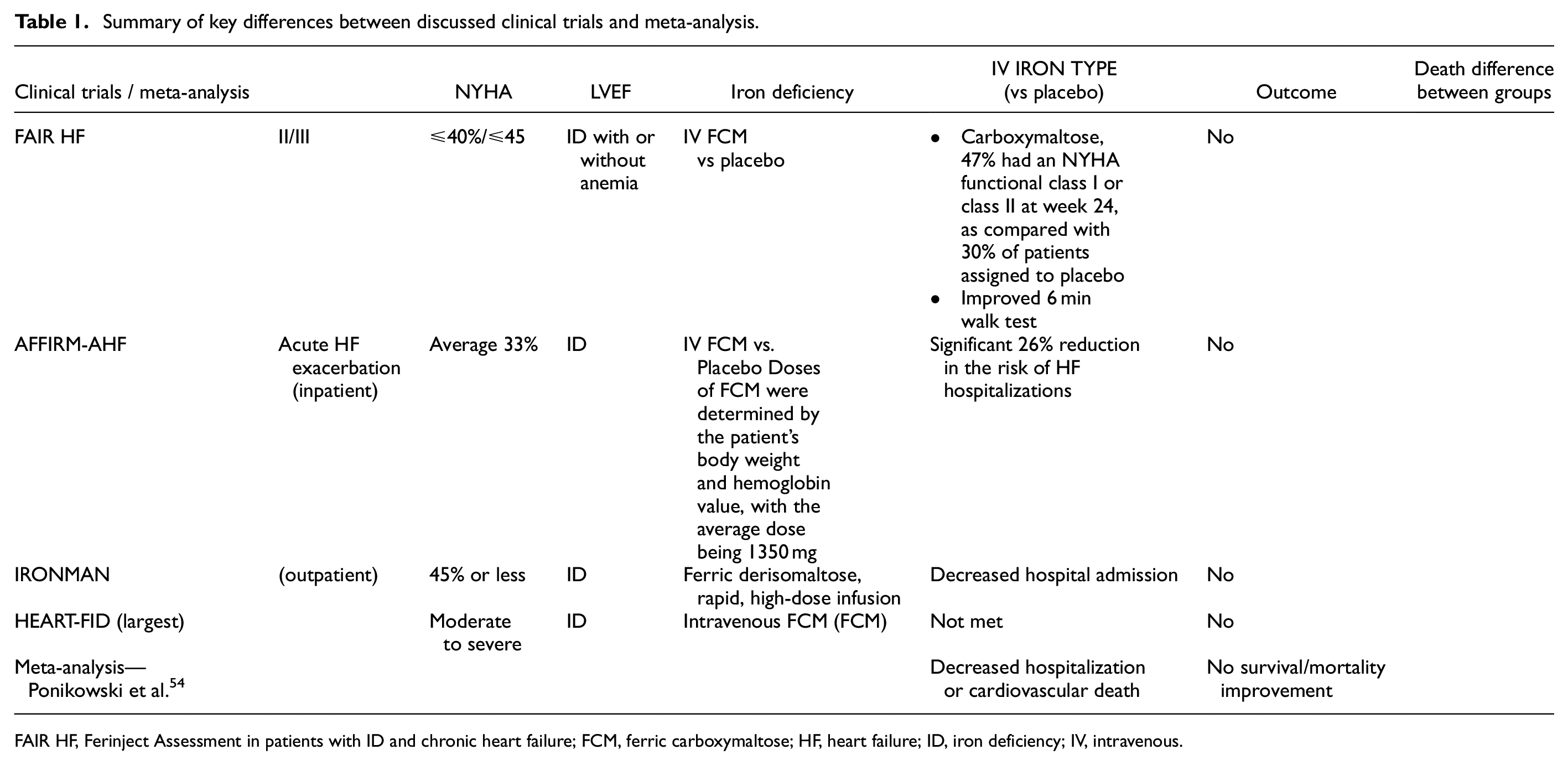
FAIR HF, Ferinject Assessment in patients with ID and chronic heart failure; FCM, ferric carboxymaltose; HF, heart failure; ID, iron deficiency; IV, intravenous.
Discussion
The broad review regarding iron homeostasis, overload, deficiency, and overall regulation demonstrated that iron plays a crucial role in physiological and intracellular functions. Both extremes of iron volume have portrayed dysfunctional abnormalities. Cardiac function has been found to be compromised in instances were iron is overloaded, causing oxidative stress, mitochondrial dysfunction, cardiomyopathy, conduction abnormalities, cell death, inflammation, and fibrosis.
Conversely, ID can cause direct disruption of diastolic function, lipid handling in cardiomyocytes, mitochondrial, and ER abnormalities. Studies suggest that iron-depleted cardiomyocytes exhibit reduced ATP levels and mitochondrial dysfunction, with alterations in iron-sulfur clusters affecting cellular respiration. Replenishing ferritin levels with transferrin-bound iron reversed these effects, suggesting potential cardiomyocyte recovery.
Around 45% of patients with HF have ID; if absolute, it is defined by ferritin <100 mg/L, functional will demonstrate normal ferritin and low tSAT. Oral iron supplementation has limited efficacy in HV.
IV iron therapy has shown promising results in treating ID in HF patients. Large, randomized trials and meta-analysis, such as FAIR-HF, AFFIRM-AHF, IRONMAN, and HEART-FID have demonstrated the efficacy of IV iron supplementation with IV FCM or IV ferric derisomaltose in reducing hospitalizations in patients and improving QoL with HFrEF, NYHA II–III.
However, survival and mortality have demonstrated no improvement in patients with HFrEF receiving IV iron, during acute exacerbations or during outpatient management.
The potential benefits of IV iron across the entire HF spectrum, including patients with preserved EF or asymptomatic left ventricular dysfunction, and its interaction with other HF therapies like SGLT2 inhibitors remain areas for further research.
Despite controversies, the American College of Cardiology (ACC) and European Society of Cardiology (ESC) recommend following therapeutic management for patients with ID and HF as below 55 :
Limited discussion has been held regarding oral iron therapy in patients with HF, primarily due to the lack of evidence substantiating its efficacy in this demographic. However, notable studies such as IRONOUT HF, a multicenter randomized controlled trial (RTC) comparing oral iron versus placebo in a 1:1 ratio among patients with reduced EF (<40%) and ID, revealed no significant enhancement in its primary endpoint. Notably, the prescribed regimen (150 mg orally twice daily) has now been proved to be ineffective. Furthermore, cardiovascular mortality and HF-related hospitalizations were not included as primary or secondary endpoints.
As discussed previously, although iron supplementation may not significantly impact survival rates, RTCs have indicated enhanced cardiac contractility, improved QoL, and reduced hospital admissions. Consequently, there is merit in conducting novel trials investigating HF and ID, incorporating oral iron supplementation with revised dosing schedules, including current guideline recommendations, involving daily oral iron intake below 150 mg or every-other-day. Studies support these regimens; they are well-tolerated, exhibit improved absorption rates, reduced side effects, and consequently, elevate hemoglobin levels.
Footnotes
Acknowledgements
AI-ASSISTED technology (ChatGPT) was used for proofreading, language polishing, spelling, and grammar.
Author contributions
D.O.M, O.A.F.B., B.X.M.E., M.E.T.P., E.C., and C.G.M. wrote and edited the manuscript, table, and figure. All authors read and approved the final version of the article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.