
Abstract
The FK506-binding protein (FKBP5) plays significant roles in mediating stress responses by interacting with glucocorticoids, participating in adipogenesis, and influencing various cellular pathways throughout the body. In this review, we described the potential role of FKBP5 in the pathogenesis of two common chronic liver diseases, metabolic dysfunction-associated steatotic liver disease (MASLD), and alcohol-associated liver disease (ALD). We provided an overview of the FK-binding protein family and elucidated their roles in cellular stress responses, metabolic diseases, and adipogenesis. We explored how FKBP5 may mechanistically influence the pathogenesis of MASLD and ALD and provided insights for further investigation into the role of FKBP5 in these two diseases.
Introduction
Intracellular homeostasis plays a crucial role in regulating the stress response and maintains a dynamic equilibrium in the face of internal or external challenges. 1 These mechanisms involve a complex physiological response that encompasses multiple genetic factors and enables the sensing, integration, and adaptation to environmental changes. 1 FK506-binding protein 51 (FKBP51 or FKBP5), an important modulator of stress responses, critically contributes to the maintenance of metabolic homeostasis by playing pivotal roles in glucose and lipid metabolism. 2 Dysfunction in FKBP5 and their signaling pathways are known contributors to the development of metabolic diseases. Studies in humans have reported associations between FKBP5 and specific disorders, including major depressive disorder, asthma, obesity, and type 2 diabetes (T2D).3–5 The tissue distribution of the FKBP5 gene corresponds to its diverse functions. FKBP5 exhibits higher expression in adipocytes,5,6 skeletal muscle, 7 and the immune system. 8 Because of its expression profile, it is not surprising that animal and cell culture studies have revealed novel and potentially significant roles of FKBP5 in mediating several metabolic pathways.2,3,9 In recent years, there has been a growing interest in exploring the potential relevance of FKBP5 in the pathogenesis of two common chronic liver diseases, metabolic dysfunction-associated steatotic liver disease (MASLD), and alcohol-associated liver disease (ALD).
MASLD and ALD are the most common causes of chronic liver diseases.10,11 An increase in the prevalence and incidence of both liver diseases contributed to a rise in the epidemic of obesity and alcohol consumption.12–14 MASLD is closely associated with obesity and the presence of metabolic syndromes in the absence of excessive alcohol use.15–17 A subset of patients with MASLD may develop a more advanced disease, metabolic dysfunction-associated steatohepatitis (MASH), a condition that can progress to advanced fibrosis and cirrhosis.18,19 ALD is an adverse consequence secondary to excessive alcohol use. ALD comprises a spectrum of histopathological changes in patients with excessive alcohol consumption, ranging from alcoholic steatosis, steatohepatitis, advanced fibrosis, and cirrhosis.20,21 Alcoholic steatosis occurs in most if not all patients who consume alcohol excessively; however, the progression of ALD to advanced stages such as alcoholic cirrhosis only develops in 15%–20% of excessive drinkers.22,23 In both MASLD and ALD, once the diseases progress to decompensated cirrhosis with complications of portal hypertension, 24 the overall prognosis and survival are poor. 25
While the specific pathways of FKBP5 in mediating the pathogenesis of MASLD and ALD are not fully elucidated, preliminary studies have indicated a potential association. In this review, we explored and described the involvement of FKBP5 in these liver diseases to gain insights into their complex mechanisms.
Overview of FKBP5 and its role in cellular stress responses
FKBP5 is encoded by the Fkbp5 gene which is approximately 155 kb with 13 exons. 26 The gene is located on the short arm of chromosome 6 (6p21.31). 5 FKBP5 is expressed in multiple tissues such as liver, muscle, adipose tissues, and bone marrow myeloid cells. 26 FKBP5 is an immunophilin due to its ability to bind immunosuppressive drug, FK-506. 27 It comprises an FKBP-type peptidyl-prolyl cis-trans isomerase (PPIase) domain, an FKBP-like domain, as well as a three-unit repeat of the tetratricopeptide repeat (TPR) domain, which enables the protein to act as a co-chaperone that changes folding and activity of other proteins.3,5,27 A comprehensive review of the basic structure of FKBP5 is described elsewhere. 3 The ability of FKBP5 to bind to heat shock protein 90 (HSP90) and other co-chaperones of the steroid receptor (SR) complex underlies its important function in stress regulation. 5 HSP90 interacts directly with FKBP5 by binding to the TPR domain; cortisol binds to glucocorticoid receptors (GRs) in the cytosol where it then binds to chaperone complexes which hold both HSP90 and FKBP5 28 (Figure 1). One of the key pathways of FKBP5-mediated cellular response is its inhibitory effect on GR signaling. 5 Glucocorticoids (GCs) are steroid hormones that bind to the GR to exert their broad physiological effects. 29 GCs are primarily synthesized in the adrenal cortex under the regulation of the hypothalamic–pituitary–adrenal (HPA) axis. 29 Under unstressed conditions, GCs are released from the adrenal cortex in the circulation in a biphasic pattern, with the peak in the morning. 29 Under physiological or cellular stresses, the HPA axis is activated with the release of corticotropin-releasing hormone from the hypothalamus to induce the release of circulating adrenocorticotrophic hormone (ACTH) from the pituitary gland. The ACTH then stimulates the adrenal gland to synthesize and secrete GC hormones (cortisol). 29 At the cellular level, GCs enter the cytosol and stimulate the GR complex. Upon binding with the FKBP5 protein, it results in a reduction in the affinity of GCs to the GR and hinders the translocation of GR into the nucleus. 5 Mechanistically, FKBP5 exerts its effects by attenuating the interaction between the GR complex and the transport protein dynein, thereby impeding GR nuclear translocation, and reducing the GR-dependent transcriptional activity.5,30 However, once FKBP5 binds to the GR complex, it is subsequently displaced by another FK binding protein, FKBP4, which recruits dynein into the GR complex and allows cellular translocation and transcription to proceed. 31 In the nucleus, GR binds to the GC response element sequences and regulates the transcription of the target genes in a tissue-specific manner. 5 Such activation also triggers multiple negative feedback loops to restrain the activity of the HPA axis, consequently regulating the release of the GCs from the adrenal gland. 32 Of importance, GR activation also leads to an increase in FKBP5 transcription and translation 26 (Figure 1). In cultured human cells, the expression of the FKBP5 gene is significantly upregulated by GCs.26,33 The newly synthesized FKBP5 can subsequently inhibit the GR activity through binding with the GR complex while modulating several molecular signaling pathways (discussed below) to regulate cellular functions. 5
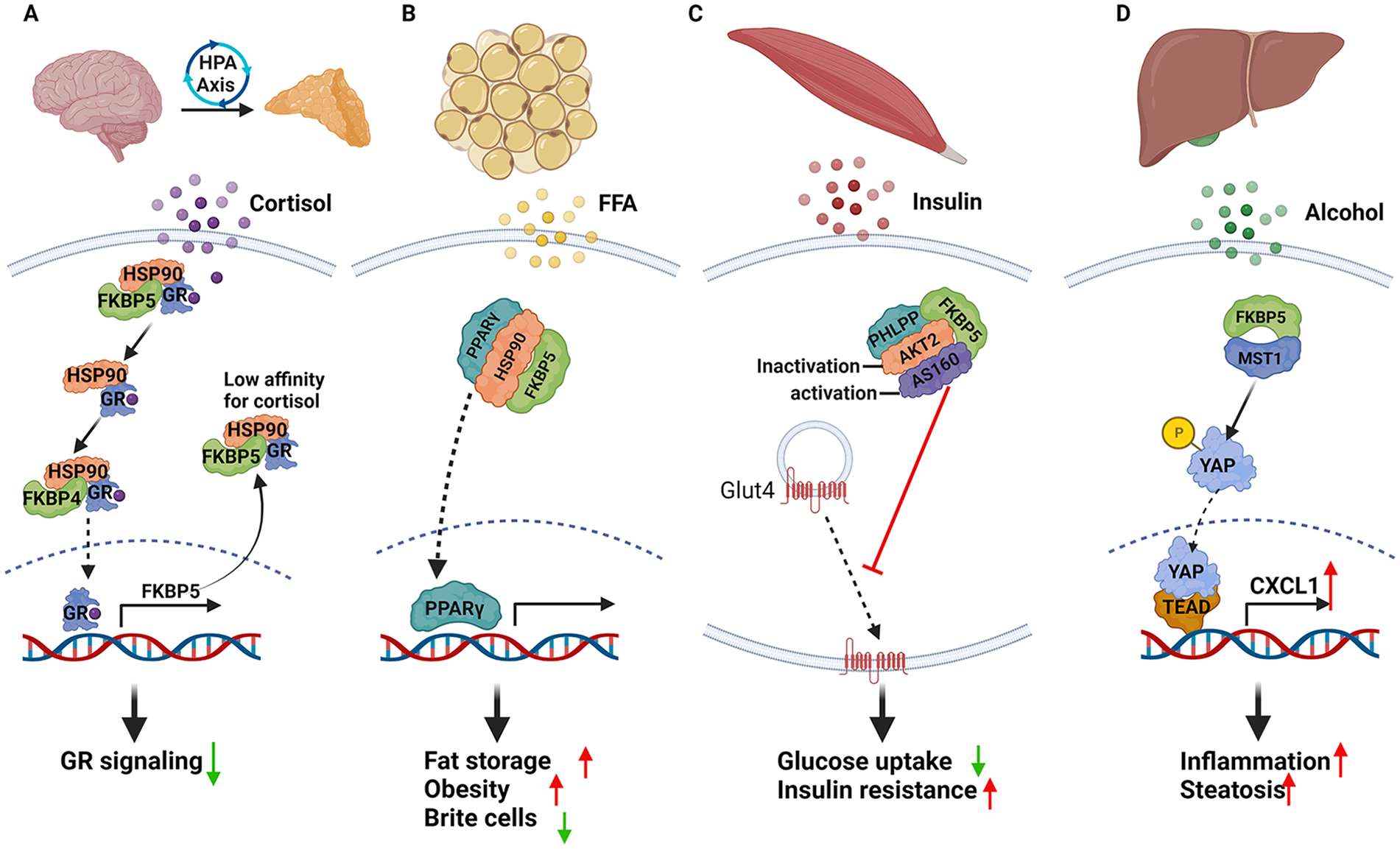
Schematic diagram of FKBP5 and its roles in mediating cellular function in different organs. (a) As a co-chaperone protein, FKBP5 can bind with other proteins to enact downstream signaling and mediate cellular function. The FKBP5 TPR domain interacts with the highly conserved HSP90 MEEDV motif3,34 and is further involved in regulating the GR and other SR signaling.3,5,35 Once formed, the FKBP5–HSP90–GR/SR complex exerts the effects on steroid hormone binding, nuclear translocation, and activation of the target genes. 5 (b) In adipose tissue, FKBP5 controls adipogenesis through its effect as a positive regulator of PPARγ, a key transcription factor in the adiopogenic process and as a negative regulator of GR through AKT and P38 MAP kinase. 6 (c) In skeletal muscle, FKBP5 regulates phosphorylation of AKT2 and its downstream AS160, a key signaling protein involved in insulin-stimulated glucose transport in skeletal muscle. The deficiency of FKBP5 improves insulin resistance and increases cellular glucose uptake. 7 (d) In alcoholic liver, the FKBP5 expression is upregulated by alcohol. FKBP5 interacts with MST1 affecting its ability to phosphorylate YAP, thus translocating YAP from the cytosol to the nucleus leading to TEAD1 activation. Activation of TEAD1 increases the expression of its target gene CXCL1, a neutrophil recruiter, causing hepatic inflammation. 36
FKBP5 and molecular signaling pathways in mediating cellular function
As a co-chaperone protein, FKBP5 can bind with other proteins to enact downstream signaling and mediate cellular function. 5 FKBP5 is known to be a co-chaperone of the HSP90 complex, a ubiquitous chaperone protein in most cells. 3 The FKBP5 TPR domain interacts and binds with the highly conserved HSP90 MEEDV motif.3,34 The binding between FKBP5 and HSP90 is essential in regulating the GR and other SR signaling, such as progesterone receptor (PR), androgen receptor (AR), and mineralocorticoid receptor (MR).3,5,35 Once formed, the FKBP5–HSP90–GR/SR complex exerts effects on steroid hormone binding, nuclear translocation, and activation of the target genes. 5 The affinity of the SR to bind to the TPR domain of FKBP5 and HSP90 is varied. 35 GR and PR display the most sensitive and apparent reaction to the TPR domain, whereas AR’s activity is moderately impaired by most cofactors. 35 Similar with its consequence on the GR, FKBP5 exerts an inhibitory effect on the PR and MR activities.5,37,38 Conversely, the AR is activated by the FKBP5 chaperone complex.39,40
FKBP5 can also bind and alter the phosphorylation state of its target proteins. Glycogen synthase kinase 3β (GSK3β), a serine–threonine kinase, regulates energy homeostasis and plays an important role in the pathogenesis of many diseases.41,42 The activity of the GSK3β depends on its phosphorylated state; the phosphorylation at serine 9 inhibits its enzymatic activity and activates the downstream signaling pathways. 34 The phosphorylation of GSK3β is tightly controlled by the upstream kinases such as AKT or protein kinase B or Cyclin-dependent kinase 5 (CDK5) and protein phosphatases such as protein phosphatase 1 and 2A.43–45 FKBP5, through its PPIase domain, binds with GSK3β and modulates the composition of the GSK3β’s complex by associating with the phosphatase PP2A and the CDK5. 41 As a result, FKBP5 increases the phosphorylation of GSK3β at the serine 9 site, inhibits the GSK3β activity, and activates the GSK3β’s downstream targets such as β-catenin and Tau,41,45 influencing various cellular processes.
FKBP5 negatively regulates all three isoforms of the serine/threonine protein kinase AKT (AKT1, AKT2, and AKT3). 7 The activity of AKT is regulated by its phosphorylation and FKBP5 acts as a scaffolding protein to modulate of AKT’s phosphorylated status. Specifically, FKBP5 functions to recruit protein phosphatase, pleckstrin homology (PH) domain, and leucine-rich repeat protein phosphatases and facilitate its dephosphorylation of AKT. 46 AKT is a critical regulator involved in insulin signaling and glucose metabolism,47,48 cell cycle,49,50 cell survival,51,52 glycogen synthesis, 43 and autophagy.53,54 By regulating the phosphorylation of the AKT, FKBP5, therefore, affects numerous cellular functions that are vital for cell growth, metabolism, and survival.
NF-κB, the nuclear factor kappa-light-chain-enhancer of activated B cells, is a protein complex with essential roles in controlling multiple cellular processes such as inflammation, proliferation, maturation, differentiation, survival, and apoptosis.55–57 It is ubiquitously found and acts as a stress response to external stimuli in multiple cell types. 58 NF-κB plays an important role in the immune response process 59 and is associated with inflammatory and metabolic diseases. 60 The NF-κB subunits, comprising the NF-κB complex, reside in the cytosol during an inactive state. 61 Upon activation during cellular stress, it leads to an interaction with the IκB kinase (IKK) complex resulting in the phosphorylation of IκB and its degradation. 61 The degradation leads to the nuclear translocation of the remaining NF-κB dimer, where it binds to the DNA consensus sequence and activates the transcription of its target genes. 61 FKBP5 was described as a regulator of NF-κB signaling. Several members of the IKK complex, IKKα, IKKβ, and IKKγ, have been identified to interact with FKBP5 involved in NF-κB signaling activation.62–65 FKBP5 could positively affect the formation and activation of the IKK complex and further promote NF-κB signaling.3,5,66
The Hippo pathway is an important regulator of cellular differentiation, proliferation, and homeostasis. 67 The pathway responds to cellular stress by the activation of several upstream serine/threonine kinases, such as MST1/2 and large tumor suppressor 1 and 2 (LATS1/2), leading to the phosphorylation of the protein targets Yes1 associated transcriptional regulator (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), which promotes their degradation and cytosolic retention and inhibits the activation of TEAD-mediated transcription. 67 The alterations of the Hippo signaling pathway are associated with various diseases and metabolic dysfunction.68,69 FKBP5 has been identified to interact with YAP upstream kinase, MST1, affecting its ability to phosphorylate YAP 36 (Figure 1). By inhibiting YAP phosphorylation, FKBP5 promotes YAP nuclear translocation and enhances TEAD activation through its role as TEAD transcriptional co-activator. 70
In summary, FKBP5 can bind to proteins in multiple pathways which regulate various cellular functions. The binding, in some instances, affects the post-translational modification (i.e., phosphorylation) of the protein targets and dysregulates the downstream cellular signaling pathways. The alterations of FKBP5, therefore, may lead to diverse consequences affecting cellular and tissue dysfunction.
FKBP5 and its function in adipose tissue
As diverse signaling pathways are involved in the regulation of homeostatic systems, FKBP5 plays a significant role in mediating the crosstalk between stress and metabolic disorders. Several studies suggested the important role of FKBP5 in regulating whole-body energy metabolism, based on its expression primarily in the metabolically active tissues, such as the liver, adipose tissue, small intestine, ovary, thymus, heart, and kidney.9,36,71,72 Adipose tissue serves as the energy storage and thermal regulation. It is also an endocrine organ with the ability to secret adipokines, a group of active molecules involved in the regulation of metabolic homeostasis, immune responses, and energy balance.73,74 The adipose tissue can be categorized based on its distribution, subcutaneous, and visceral adipose tissues. 75 Central rather than peripheral adiposity is a risk factor for metabolic diseases.75,76 The limited expandability of the adipose tissue in the subcutaneous area causes an unacceptable adipose cell expansion and insulin resistance.75,77 The expansion of adipose tissue by increasing adipogenesis distributes excess fat between newly differentiated adipocytes and decreases the number of inflammatory cytokines producing adipocytes. 75 Adipogenesis is a multi-step and tightly regulated process when the pluripotent mesenchymal stem cells (MSCs), upon stimulation, commit to the changes into the adipocyte lineage. 75 The initial phase is the conversion of the MSCs into pre-adipocytes, which then differentiate into mature adipocytes with specific functions, such as lipid storage, lipid synthesis, insulin sensitivity, and the secretion of adipokines. The defect in the process leads to adipose tissue dysfunction and insulin resistance. 75
One of FKBP5’s functions in mediating metabolic processes is its effect on adipocyte differentiation. 74 During the clonal expansion phase of adipocyte differentiation in 3T3-L1 cells, the expression of FKBP5 transcript and its protein level exhibit similar temporal kinetics. 78 Its expression is essential to cellular adipogenesis for the transformation of pre-adipocytes into mature, lipid-bearing cells.6,9 FKBP5 expression level increases during 3T3-L1 preadipocyte differentiation and it rapidly shuttles from mitochondria to the nucleus at the onset of adipogenesis, the process regulated by the second messenger cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA) signaling pathways. 74 When the adipogenesis is activated, cAMP–PKA regulates the rapid nuclear translocation of FKBP5, which progressively increases its interaction with GR and restrains its transcriptional capacity. 74 Knockdown of Fkbp5 accelerates adipogenesis, illustrated by an increase in the level of adipokines such as adiponectin and resistin. 74 Conversely, ectopic expression of FKBP5 impedes adipogenesis with a significant reduction in the percentage of cells with the capacity to differentiate into adipocytes. 74 In the absence of FKBP5, the ability of 3T3-L1 and mouse embryonic fibroblasts (MEFs) to differentiate into lipid-laden cells is inhibited in parallel with the reduction in the enzymatic activity of fatty acid synthase and other lipogenic genes as well as the deceased expression of the cluster of differentiation 36, a membrane protein responsible for fatty acid uptake. 6 Mechanistically, FKBP5 controls adipogenesis through its effect as a positive regulator of peroxisome proliferator-activated receptor gamma (PPARγ), a key transcription factor in the adipogenic process, and as a negative regulator of GR through AKT and P38 mitogen-activated protein (MAP) kinase. 6 In one study using hypoxia-induced adipogenesis in the MEFs, the loss of FKBP5 significantly prevents adipogenesis and reduces the expression of adipogenesis-related genes, including those involved in lipogenesis, lipolysis, and energy metabolism. 79 Taken together, FKBP5 is a negative regulator of adipogenesis.74,79
The effect of FKBP5 in human adipose tissue is studied to explore the effects of adipose tissue insulin resistance induced by GC. 72 Paired biopsies from abdominal subcutaneous and visceral adipose tissues were obtained from patients undergoing elective surgery. 72 The protein expression of FKBP5 is increased in a dose-dependent manner when both subcutaneous and visceral adipose tissues are treated with dexamethasone. Notably, the increase in FKBP5 protein was significantly higher in visceral adipose tissue treated with dexamethasone compared to subcutaneous tissue. 72 The change in FKBP5 gene expression induced by dexamethasone correlated negatively with several metabolic parameters, such as serum insulin, homeostatic model assessment for insulin resistance (HOMA-IR), and glycosylated hemoglobin, and positively with HDL-cholesterol in subcutaneous, but not in visceral adipose tissue. 72 Mechanistically, FKBP5 was found to affect the glucose uptake in human adipocytes, primarily from the visceral adipose tissue. Dexamethasone treatment led to decreased both basal and insulin-stimulated glucose uptake in the adipocytes isolated from both subcutaneous and visceral adipose tissues. 72 Patients with higher endogenous FKBP5 gene expression in the visceral adipose tissue showed a reduction in the insulin effects on the glucose uptake in adipocytes from both subcutaneous and visceral tissues. 72 However, the higher endogenous Fkbp5 gene expression in the subcutaneous adipose tissue did not exert any insulin effects in the adipocytes. 72 In another study, there was a trend in an increase in FKBP5 gene expression in the subcutaneous adipose tissue in patients with T2D when compared to healthy controls. 9 The expression of FKBP5 positively correlated with the markers of insulin resistance and negatively associated with the oral glucose tolerance test (OGTT)-derived insulin sensitivity indices. 9 FKBP5 gene expression levels were also associated with genes regulated by the GR, with a positive association with GR, and a negative association with HSP90B1. 9 Furthermore, FKBP5 expression in the subcutaneous adipose tissue was negatively associated with genes involved in lipolysis and adipogenesis. 9 These studies illustrate the important role of FKBP5 in the adipose tissues in mediating adipogenesis, glucose and lipid metabolism, and metabolic dysfunction.
FKBP5 and its association with several metabolic diseases
DNA methylation is an epigenetic modification of DNA that is important for the normal regulation of transcription, embryonic development, genomic imprinting, genome stability, and chromatin structure. Aberrations in the machinery responsible for DNA methylation patterns and chromatin structure also contribute to human disease. 80 Methylation of the FKBP5 gene has been reported to be associated with metabolic dysfunction.66,81 Methylation of FKBP5 was analyzed in gluteal and abdominal subcutaneous adipose tissue from obese and normal weight women. 82 For FKBP5 intron 7, the methylation levels of CpG542 (137,847 bp from transcriptional start site (TSS) and CpG543 (137,872 bp from TSS) were approximately 10% higher in obese compared to normal weight patients in both GSAT and ASAT. 82 CpG542 and CpG543 were positively associated with the measures of adiposity such as waist circumference and body mass index in both GSAT and ASAT, the markers of insulin resistance (such as HOMA-IR and fasting insulin), and systemic inflammation. 82
As a stress response protein, FKBP5 appears to be a molecular mediator linking stress with metabolic phenotypes. 83 A comprehensive review of the role of FKBP5, stress, and metabolic disorders has been described elsewhere. 2 Such association would be predicted that individuals carrying a high percentage of CpG methylation in FKBP5 DNA are more susceptible to metabolic diseases when exposed to external stimuli or stresses. Single nucleotide polymorphisms (SNPs) are variations in the genome, and they are responsible for rendering susceptibility to specific diseases.22,84–86 Of interest, there is also an association between the SNPs in the FKBP5 region and metabolic phenotypes. The rs2817056 and rs2395635 are associated with T2D and impaired 2-h OGTT, respectively. In addition, three SNPs, rs1334894, rs9380525, and rs9368881, are associated with the level of high-density lipoprotein (HDL) -cholesterol, while six SNPs are linked to serum triglycerides (rs9394309, rs10947563, rs7763535, rs2395635, rs6912833, and rs4713904). 72 Another piece of evidence confirming the role of FKBP5 variants in metabolic regulation comes from studies investigating the association of the FKBP5 rs1360780 genotype on weight loss outcomes after bariatric surgery.87,88 Patients with the T allele of the rs1360780 polymorphism, the variant with an increase in FKBP5 responsivity, have approximately 20% less excess weight loss and 10% less total weight loss compared with those of the alternate C allele during the follow-up.83,88
In another study, the Fkbp5 knockout mice fed with a standard chow diet exhibited a modest reduction in body weight, adiposity, and an increase in lean mass compared to wild-type controls. 7 Similar to the aforementioned study, 89 these mice were resistant to high-fat diet-induced weight gain and adiposity and exhibited an improvement in glucose tolerance and an increase in insulin sensitivity primarily in the skeletal muscle without alterations in circulating insulin level. 7 Mechanistically, FKBP5 was found to be a crucial regulator of glucose disposal through the regulation of alpha serine/threonine kinase 2 (AKT2) and its downstream AS160 (AKT substrate of 160 kDa), a key signaling protein involved in insulin-stimulated glucose transport in skeletal muscle 7 (Figure 1). The lack of FKBP5 led to increased phosphorylation of AKT2, AS160, p70S6K, and GLUT4 (Glucose transporter type 4) translocation in the skeletal muscle. 7 The FKBP5 deletion also affected the gut microbiota and prevented the development of obesity and metabolic phenotypes. 90
FKBP5 and metabolic dysfunction-associated steatotic liver disease
A recent study discovered a dysregulation of FKBP5 gene expression associated with insulin resistance in induced pluripotent stem cell-derived hepatocytes from MASLD patients. 91 Similarly, another study found an upregulation of FKBP5 in the liver affected by MASH. 92 As previously mentioned, FKBP5 induction can serve as a surrogate marker for GR activation and elevated GC levels are associated with metabolic syndrome and obesity. 93 The potential relationship between FKBP5, obesity (a common metabolic disorder in MASLD), and the HPA axis has been examined in a nutritional deprivation model. Nutritional deprivation leads to an increase in the hypothalamic FKBP5 transcript and a reduction of negative feedback within the HPA axis. 93 Overexpression of hypothalamic FKBP5 using adeno-associated virus results in elevated serum cortisone, increased body weight, and impaired glucose tolerance in mice fed with a high-fat diet compared to mice receiving control vectors. 93 By contrast, mice lacking Fkbp5 demonstrate leanness under a normal chow diet.2,94 Moreover, Fkbp5 knockout mice fed with a high-fat diet for 4 weeks are resistant to weight gain, adiposity, and hepatic steatosis. 89 Lower plasma lipids and improved insulin sensitivity are also observed in mice lacking Fkbp5 fed with a high-fat diet. 89 These mice also exhibit higher respiratory exchange ratios, energy expenditure, and oxygen consumption compared to wild-type mice fed with a high-fat diet. 89 The protective effect of Fkbp5 deficiency on hepatic steatosis may be attributed, in part, to its role in enhancing energy expenditure and improving insulin sensitivity by repressing PPARγ activity. 89 In addition, because of the role of FKBP5 in mediating adipogenesis, Fkbp5 knockout mice fed with a high-fat diet show resistance to white adipose tissue expansion while maintaining brown adipose tissue. In another study, FKBP5 led to a worsening of MASH through its interaction with the GR. 92 These effects were ameliorated by intraperitoneal injections of Honokiol, which activated the GR and subsequently induced MIG6, a tumor suppressor protein. Since FKBP5 acts as a GR cochaperone, the researchers suggest that FKBP5 may exacerbate nonalcoholic liver diseases by displacing the GR complex, activating the epidermal growth factor receptor (EGFR) pathway, and promoting tumor progression. These studies not only highlight the significant role of FKBP5 in the MASLD pathogenesis but also identify it as a potential target for future drug intervention.
Recent genome-wide association studies (GWAS) investigating MASLD have not identified any significant signals related to FKBP5,95,96 suggesting that the gene may not play a prominent role in the genetic predisposition to MASLD. However, GWAS encompass numerous genetic loci: most disease-associated variants have small effect sizes. Identifying core genes directly related to the disease with clear biological relevance can be challenging. While GWAS studies may not provide strong support for the involvement of FKBP5 in MASLD, the findings from the mechanistic studies outlined above suggested its role in MASLD pathogenesis.
FKBP5 in alcohol drinking and alcohol-associated liver disease
Excessive alcohol use (EAU) is one of the most significant risk factors for health problems. 97 Genetic variants of FKBP5 are correlated with an increased risk of alcohol dependence and EAU.98–101 Moreover, FKBP5 is related to alcohol withdrawal severity. One study revealed that minor alleles of rs3800373 (G), rs9296158 (A), rs1360780 (T), and rs9470080 (T) in FKBP5 were significantly associated with lower Clinical Institute Withdrawal Assessment for Alcohol-Revised scores, whereas the minor alleles of rs3777747 (G) and rs9380524 (A) were associated with higher scores in 399 alcohol-dependent inpatients with alcohol consumption. 102 The haplotype-based analyses also showed an association with alcohol withdrawal severity. 102 Fkbp5 KO mice showed significantly greater handling-induced convulsions during withdrawal from chronic alcohol exposure compared with wild type (WT) controls. 102 Another study also found polymorphisms (SNPs) in FKBP5 are associated with alcohol drinking in humans. 103 In mice, Fkbp5 KO mice exhibited an increase in alcohol consumption, and higher blood alcohol concentration was found in these mice after 3 h of alcohol access. 103 A recent study reported that FKBP5 inhibitor reduced alcohol preference in stressed male and female mice. 104
ALD is a complex disorder that develops as a consequence of EAU. Recently, our group identified an FKBP5–YAP–TEAD1–CXCL1 axis in the pathogenesis of ALD. 36 An increase in the expression of FKBP5 at the transcript and the protein levels is also found in the liver of mice fed with alcohol and in patients with ALD. 36 The loss of FKBP5 protects against alcohol-induced hepatic steatosis and reduces hepatic neutrophil infiltration and the level of hepatic inflammatory cytokines. 36 In alcohol-fed mice, the upregulation of FKBP5 expression by alcohol is secondary to the downregulation of methylation level at its 5′ UTR promoter region. 36 An increase in FKBP5 expression leads to an induction of transcription factor TEA Domain Transcription Factor 1 (TEAD1) through the Hippo signaling pathway. FKBP5 interacts with mammalian Ste20-like kinase 1 (MST1) affecting its ability to phosphorylate YAP, thus translocating YAP from the cytosol to the nucleus leading to TEAD1 activation. Activation of TEAD1 increases the expression of its target gene C-X-C Motif Chemokine Ligand 1 (CXCL1), a neutrophil recruiter, causing hepatic inflammation 36 in the mouse model (Figure 1). These findings highlight the intricate molecular pathways involving FKBP5, TEAD1, and CXCL1 that contribute to the development of hepatic inflammation in ALD. Further research in this area can provide deeper insights into the underlying mechanisms and potentially identify FKBP5 as a therapeutic target for ALD.
Conclusion and future perspective
The expression of FKBP5 is widely distributed throughout various tissues in the human body. Alterations in FKBP5 expression can result in downstream effects and contribute to organ dysfunction. This review focuses on the role of FKBP5 in cellular stress response and its function as a chaperone protein in mediating molecular signaling pathways. We provided an overview of its involvement in several metabolic pathways and specifically addressed its implications in metabolism-associated diseases. The selective FKBP5 ligands SAFit1 and SAFit2 have been employed in several in vivo disease models as selective pharmacological inhibitors of FKBP5. 2 Chronic treatment with SAFit2 has similar effects to that of FKBP5 deletion on the restoration of metabolic function and improving glucose tolerance. 7 Further studies investigating the potential relevance of FKBP5 to metabolic diseases such as MASLD and ALD would be highly beneficial. There is still much to uncover regarding FKBP5’s involvement in the pathogenesis and progression of these diseases. Conducting additional investigations can provide valuable insights into the specific mechanisms by which FKBP5 contributes to metabolic dysfunction and organ damage in MASLD and ALD.
Footnotes
Roles of authors
JM and ZY searched for relevant articles and drafted the manuscript. NH, HG, and YJ provided critical comments on the draft, and ZY and SL reviewed and finalized the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: ZY is supported by NIH K01AA26385, R01AA030993, Indiana University Research Support Fund Grant (IU RSFG), the Ralph W. and Grace M. Showalter Research Trust and the Indiana University School of Medicine, and Indiana Institute for Medical Research (IIMR) and the Central Society for Clinical and Translational Research (CSCTR); JM is supported in part by NIH K99AA031067 and The Indiana Clinical and Translational Sciences Institute postdoctoral challenge grant, SL is supported in part by R01 AA025208, U01 AA026917, UH2/UH3 AA026903, R01AA030312, U01AA026817, VA Merit Award 1I01CX000361, and Dean’s Scholar in Medical Research, Indiana University School of Medicine.