
Abstract
Molecular investigations into polymer-surface interactions are essential for understanding and optimizing the mechanical behavior of, e.g., adhesive joints or polymer nanocomposites. In this context, metadynamics provides a powerful tool for determining the free energy of a molecular system through Gaussian sampling. While the literature demonstrates that metadynamics is well suited for adherend–adhesive systems, there is little discussion on the choice of the metadynamics parameters and their impact on the resulting free energy. This contribution provides a step-by-step methodology for determining the parameters of metadynamics, specifically the Gaussian’s depth, width, and deposition time, as well as the convergence. To illustrate this process, we utilize a simple coarse-grained system made up of a rigid substrate and either an epoxy or hardener molecule. Once we establish the optimal metadynamics parameters, we examine the effects of non-bonded polymer-surface interactions, which are modeled using 12-6 Lennard–Jones potentials, on the resulting free energy surface. As expected, we find that the distance parameter
1. Introduction
Structural adhesives are high-performance bonding agents that provide strong, durable connections between various materials and are thus a key technology for cost-efficient and sustainable engineering solutions. Compared to conventional fastening methods, such as riveting, bolting, or welding, they offer a better strength-to-weight ratio and allow for bonding dissimilar materials without requiring holes [1,2]. This enables the use of high-performing fiber-reinforced polymers [3], making structural adhesive bonds widely used today in automotive [4], aerospace [5], and civil engineering [6]. The adhesive is often based on epoxy resins [7], as these are highly durable and cost-efficient [8]. Consequently, a curing process is required in which the epoxy and a hardening agent form a polymer network, which enables the load transfer. As a side effect of the curing, an interphase region forms in the adhesive near the adherend surfaces. This interphase differs from the bulk adhesive in terms of curing degree [9], glass transition [10], and mechanical properties [11,12]. Consequently, a comprehensive understanding of the interphase, particularly of its mechanical properties, is key to unlock the full potential of adhesive joints. However, this interphase, depending on material and process parameters, exhibits a thickness of only a few nanometers to several micrometers [10,13] and is therefore difficult to detect and characterize via experiments [11]. Similar interphase effects and challenges arise for other polymeric interphases, e.g., in polymer nanocomposites [14–16].
As a remedy, particle-based numerical methods, such as molecular dynamics (MD), are commonly employed since these offer the required spatial resolution to accurately model the adhesive’s microstructure [17]. All-atom MD simulates every individual atom and thus offers detailed insights into the interphase formation [18,19], the impact of different adherend surfaces [20,21], or the detrimental effect of moisture [22,23]. However, all-atom MD is too computationally expensive to simulate samples large enough to evaluate the mechanical properties. The much more efficient coarse-grained MD (CGMD) solves this issue by substituting groups of atoms by superatoms, which considerably reduces the degrees of freedom [24]. Thus, CGMD facilitates the investigation of the mechanical behavior of adhesive joints [25,26] and their interphases [27–29]. One crucial issue is the correct coarse-grained representation of non-bonded interactions, i.e., the Van der Waals and Coulomb interactions between the substrate surface and the polymer. One practical approach is to calibrate the coarse-grained potentials so that CGMD replicates the all-atom free energy surface, evaluated with metadynamics. Metadynamics [30] is a simulation method that introduces a time-dependent bias potential to a molecular system, facilitating the exploration of its free energy landscape. For example, Lau et al. [31] employ metadynamics to construct the free energy of a single DGEBA molecule detaching from a silica surface. They upscale their results by estimating the intrinsic strength at the interface via a worm-like chain-based fracture model [32] and calibrate a traction-separation law describing the epoxy-silica interface at the continuum scale. In a similar vein, Yang and Qu [33] use metadynamics to obtain the coarse-grained interactions at an epoxy-copper interface. They observe a mechanical interphase region where particle displacements differ from the bulk polymer and derive a traction-separation law from their results.
Coarse-graining is commonly used to model the mechanics of adhesive joints due to its computational efficiency [17]. However, most existing models do not feature the critical surface interactions. The literature indicates that metadynamics is an effective tool for integrating adherend–adhesive interactions into established models without a complete reparametrization. While transferability errors may arise, this metadynamics-based extension offers an efficient exploration of surface-chemistry effects, yielding valuable qualitative insights into adhesive joint behavior. However, the identification of the metadynamics parameters and their impact on the determined free energy surface often remains ambiguous in the literature. Therefore, this article uses the example of a simple coarse-grained adherend–adhesive system to illustrate the calibration of a metadynamics analysis in detail. Our research questions are: 1) How are the metadynamics parameters calibrated for an adherend–adhesive system? 2) How do the non-bonded interactions between the adherend and the adhesive influence their free energy surface? Hence, this contribution aims to deliver a reproducible and instructive protocol for conducting coarse-grained metadynamics investigations of polymer–surface interactions. By employing a minimalistic coarse-grained model, we ensure the primary focus remains on the methodology of free-energy sampling, rather than the physical interpretation of the results. In Section 2, we introduce the coarse-grained model, the basics of metadynamics, and the investigated sample setup. We then determine the Gaussian width, the time to reach convergence, and the Gaussian height in Section 3. With these optimal metadynamics parameters, we determine the free energy of the system and discuss the impact of the non-bonded substrate-polymer interactions. The methodology presented here relies on open-access tools and can be easily adapted to more complex systems, making this article a valuable starting point for future research.
2. Methods
2.1. Coarse-grained model
2.1.1. Epoxy
We perform the following investigations in coarse-grained resolution. To this end, we choose a common thermoset comprising the resin Diglycidyl ether of bisphenol A (DGEBA) and the hardener 4,4’-Diaminodicyclohexylmethane (PACM). Giuntoli et al. [34] calibrated the corresponding coarse-grained interaction potentials via the Energy Renormalization approach [5,35] with Gaussian process surrogate models [36]. They focused on reproducing the mechanical properties and obtained good agreement of the Debye-Waller factor, density, Young’s modulus, and yield stress with respect to their all-atom reference simulations. In addition, the model is computationally efficient and has thus been used to study the fracture behavior [37], impact of non-stoichiometric mixing ratios [38], and the interphase formation of adhesive joints [29]. The level of coarse-graining is visualized in Figure 1, and we provide the force fields in 4. In this study, we consider single DGEBA and PACM molecules and thus do not need to simulate the curing reactions.
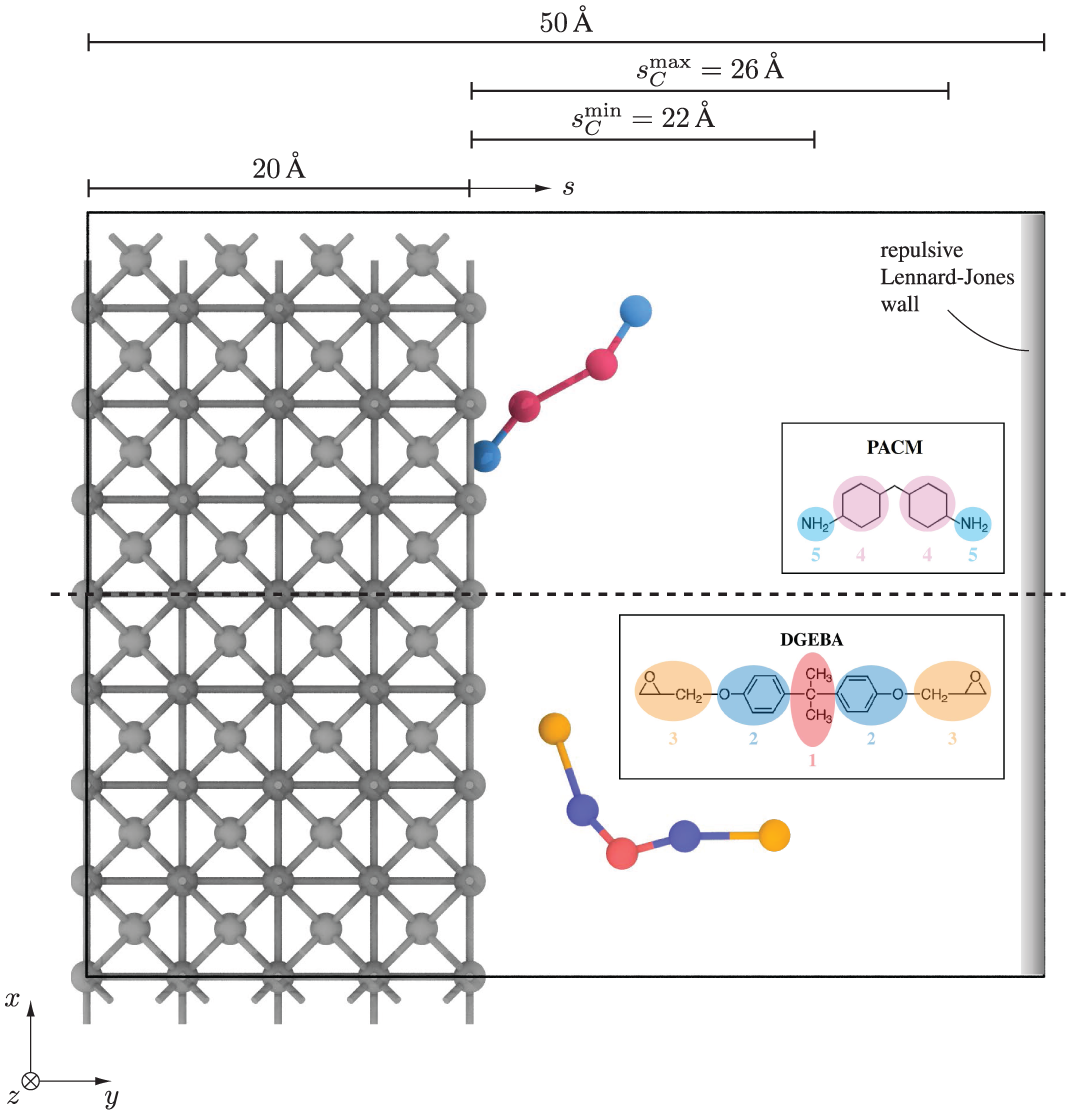
Simulation setup: rigid substrate with either PACM (top) or DGEBA (bottom) placed in simulation box, periodic boundary conditions in the x and z-directions, repulsive Lennard–Jones wall in the upper y-direction; insets visualize coarse-graining for PACM and DGEBA by Giuntoli et al. [34]; collective variable s measures the shortest distance of DGEBA’s and PACM’s center of mass from the substrate’s surface.
2.1.2. Substrate
We model the substrate, exemplarily, as a single crystal with a body-centered cubic lattice and lattice spacing
2.1.3. Transferability
Bottom-up CG models are usually calibrated based on the potential of mean force (PMF) obtained from a fine-scale, e.g., atomistic, reference simulation. The PMF is typically dependent on material composition and thermodynamic state, rendering bottom-up CG models similarly susceptible to changes in these conditions. Consequently, CG potentials are usually not transferable across different thermodynamic states (e.g., temperatures, densities, or pressures) or other local environments (bulk vs. interfacial regions) [39]. Hence, one must be extremely careful when extending an existing CG bulk model to incorporate an interface, as outlined here. The present CG model [34] employs the energy renormalization approach [35] to explicitly incorporate a wide range of curing degrees for the bulk epoxy. However, to show that the model can be extended to include an interface, it is necessary to carefully validate it using atomistic reference simulations. Because atomistic molecular dynamics (MD) simulations are usually too computationally expensive to directly analyze mechanical behavior, validation instead focuses on the structural features of the adhesive near the substrate. For example, Yang and Qu [33] derived the coarse-grained (CG) epoxy-copper interaction by detaching a single epoxy molecule in both CG and all-atom (AA) resolutions using metadynamics. They then validated their CG model by comparing the microstructural characteristics observed in densely packed AA and CG samples after curing [40]. Consequently, while calibration of adhesive-adherend interaction potentials can be performed with a single molecule, validation requires large-scale atomistic reference simulations to identify key microstructural features—an effort that is beyond the scope of this work. In general, the bi-material setup has to be considered already during the CG potential parametrization [39]. Addressing the limited transferability of coarse-grained models, novel strategies such as local density potentials [41] as well as dual [42] and microcanonical [43] approaches aim to enhance density and temperature transferability, respectively [44]. In this context, Jin et al. [45] provide a detailed review of enhanced bottom-up coarse-graining techniques and discuss transferability issues and possible remedies in depth.
2.2. Metadynamics
Metadynamics [30,46,47] identifies the free energy F of a molecular system with respect to a (small) number of collective variables s by adding external Gaussian potentials every

with Gaussian height ω and width

where

and compute the free energy via (2). In order to improve the estimation of the free energy, we compute the arithmetic average [50]

for
The sampling of
2.3. Sample configurations
Similar to other contributions [31,33], we evaluate the free energy of a single resin or hardener molecule in the vicinity of a generic substrate. To this end, we initialize a simulation box
2.4. Simulation parameters
For the MD simulations, we employ LAMMPS [51,52] with the metadynamics plugin PLUMED [48,53]. We perform all simulations at 300 K under NVT ensemble with time step size
According to [47], a successful metadynamics run requires (1) equilibrating the sample at the desired temperature, (2) choosing the collective variables, (3) setting the metadynamics parameters, and (4) ensuring convergence. Since we insert the mobile beads pre-equilibrated and the substrates are rigid, we already fulfill step 1. The collective variables have to describe the state of the system accurately, which makes their selection one of the most challenging aspects of metadynamics [47]. In our case, however, using the shortest distance between the mobile beads’ center of mass and the substrate’s surface beads, as depicted in Figure 1, appears as an obvious choice [31,33]. Due to the simple structure of the resin and hardener molecules in coarse-grained resolution, we neglect their rotation, leaving us with only one collective variable s. Using a single collective variable can lead to projection artifacts and may overlook important orientation information, potentially oversimplifying the free-energy representation of complex systems. While a two-dimensional surface could offer more detail, as demonstrated by [55], our emphasis on presenting a methodology for metadynamics sampling makes this simplification acceptable for this study. Hence, we only need to set the parameters for the actual metadynamics run, i.e., the Gaussian deposition time
By inserting pre-equilibrated DGEBA and PACM molecules and due to the straightforward selection of collective variables, we already fulfill steps 1 and 2 [47]. We address steps 3 and 4 in Section 3 by determining δs,
3. Results and discussions
3.1. Gaussian width δs
First, we determine the Gaussian width δs, which defines the resolution of the free energy surface in the collective variable space. To this end, we perform a standard (unconstrained) molecular dynamics run for 4 ns for all 30
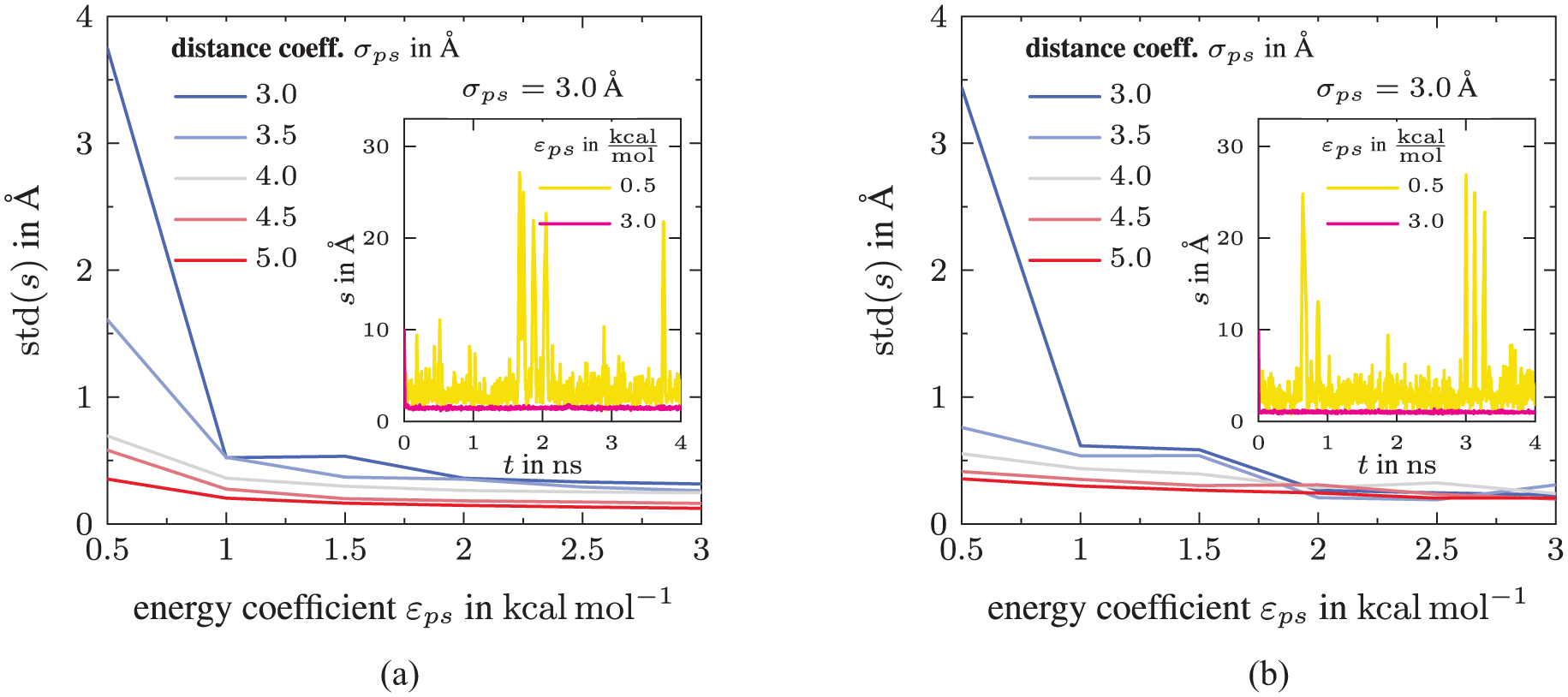
Indentification of Gaussian width δs for (a) DGEBA and (b) PACM with standard deviation
3.2. Convergence time
Second, we ensure convergence by identifying the filling time
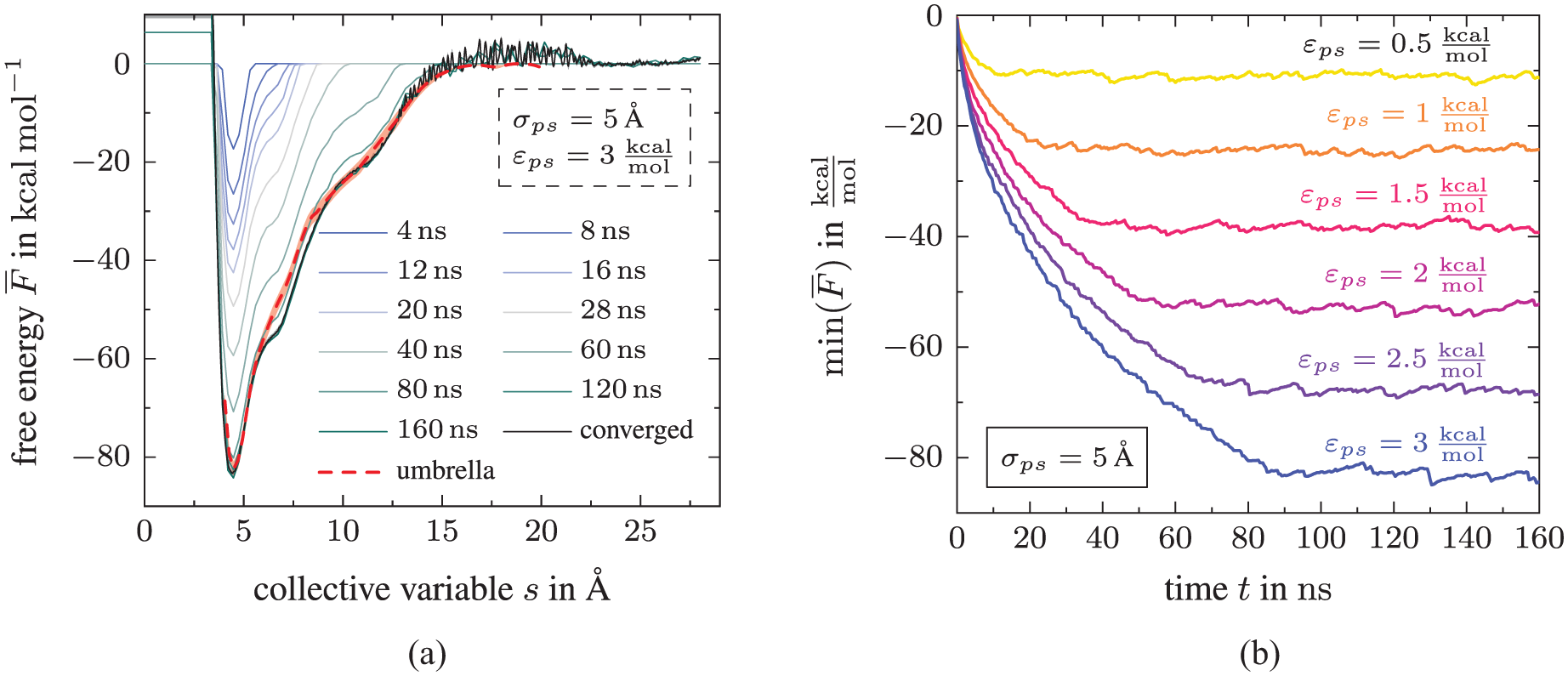
Convergence for DGEBA molecule: (a) Free energy
3.3. Gaussian height ω
According to [46], metadynamics’ accuracy is proportional to the ratio
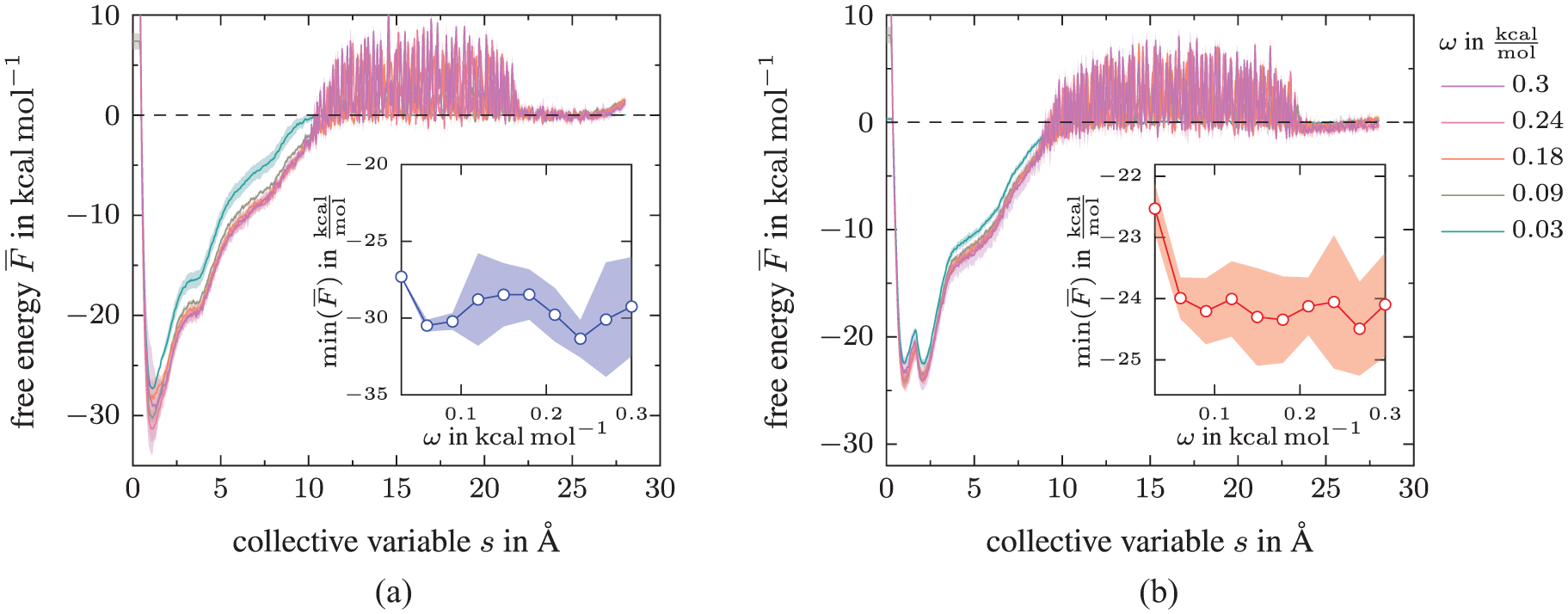
Impact of Gaussian height ω on the converged free energy landscape
3.4. Impact of non-bonded interactions
Having determined the metadynamics parameters
3.4.1. DGEBA
Figure 5(a) (left) displays the effect of the non-bonded energy parameter
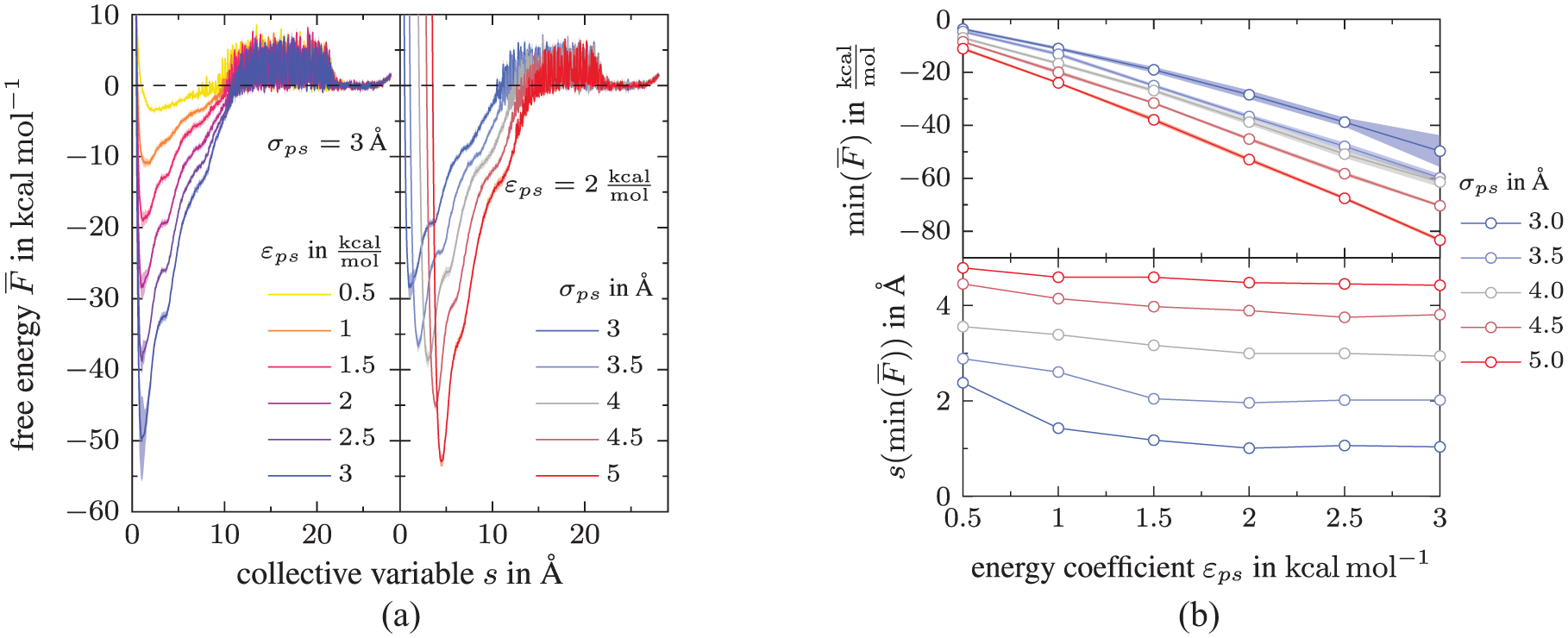
Impact of the DGEBA-substrate interactions on the free energy landscape: (a) Free energy
3.4.2. PACM
In contrast to DGEBA, the free energy of the PACM molecule in Figure 6(a) (left) features two local minima,
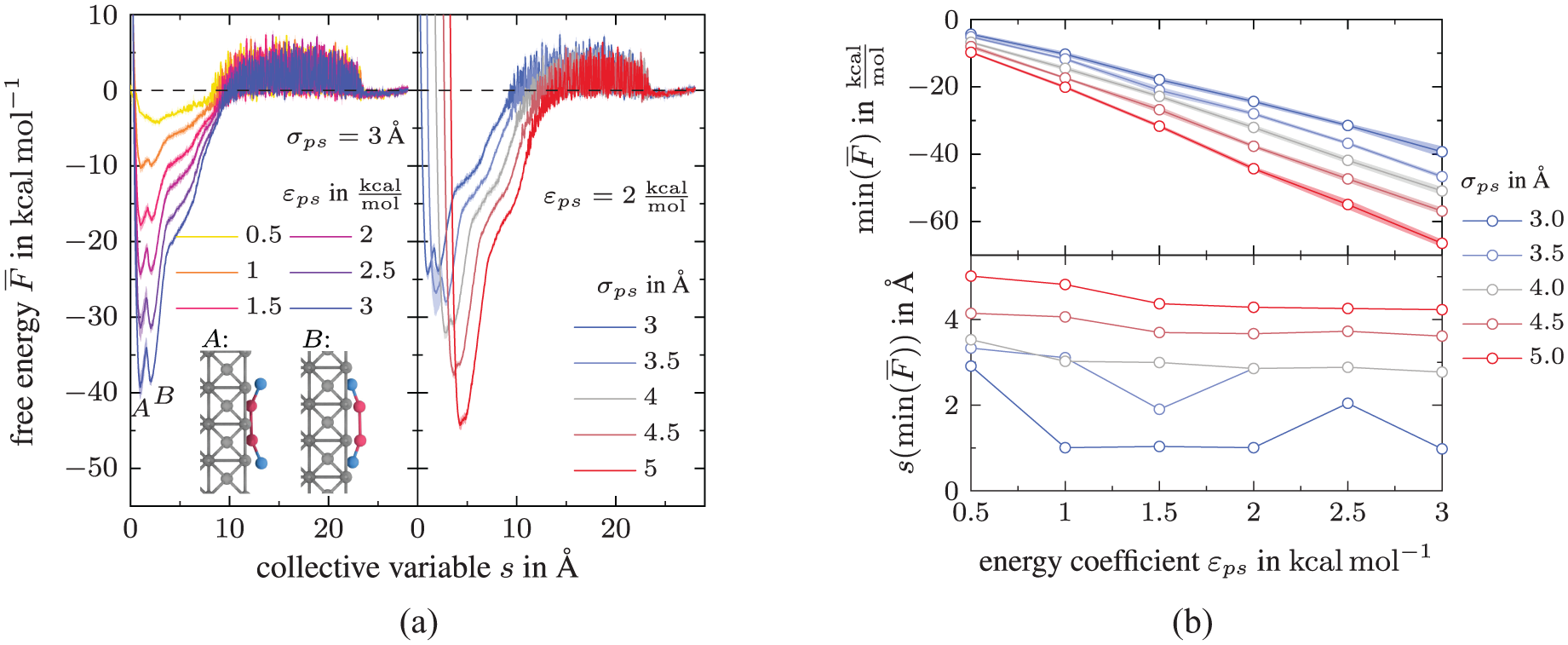
Impact of the PACM-substrate interactions on the free energy landscape: (a) Free energy
Overall, the non-bonded interactions between the substrate and resin or hardener, parametrized in the Lennard–Jones parameter
4. Conclusion and outlook
In this contribution, we demonstrate how to determine the free energy landscape of an adherend–adhesive system via metadynamics and how the free energy is affected by the non-bonded adherend–adhesive interactions. To this end, we choose a simple system consisting of one resin or hardener molecule and a substrate surface in coarse-grained resolution. We show how the metadynamics parameters, i.e., the Gaussian width, filling time, Gaussian height, and Gaussian deposition time, are calibrated step by step. The optimal Gaussian width δs results from the fluctuations of the collective variable in an unbiased MD system. We estimate the filling time
This article serves as a guide for advanced studies, and the methodology presented can be easily applied to more complex systems. We exclusively use open-source software and provide all the scripts utilized in this research via Zenodo [61], making this study a straightforward introduction to metadynamics in the context of adhesives. In addition, we intend to utilize the determined free energy dependencies of
Simulation software and data
We employ LAMMPS (version 12 June 2025) [51,52] and the open-source, community-developed PLUMED library (v2.9.4) [48,53] for the MD and metadynamics simulations performed on the High-Performance-Computing (HPC) parallel CPU cluster Fritz of the Erlangen National High Performance Computing Center (NHR@FAU) of “Regionales Rechenzentrum Erlangen (RRZE).” We used for the evaluation, data processing and visualization of the simulations mainly MathWorks MATLAB R2023b and OVITO [62], as well as the WHAM algorithm [59]. We provide the data published in this contribution via Zenodo [61].
Footnotes
Appendix 1
Acknowledgements
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: M. Ries is funded by the DFG - 377472739 (Research Training Group GRK 2423 ‘Fracture across Scales - FRASCAL’). The author gratefully acknowledges the scientific support and HPC resources provided by the Erlangen National High Performance Computing Center (NHR@FAU) of the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) under the NHR project b275dc “Molecular Modeling of Adhesive Joints”. NHR funding is provided by federal and Bavarian state authorities. NHR@FAU hardware is partially funded by the German Research Foundation (DFG) - 440719683.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.