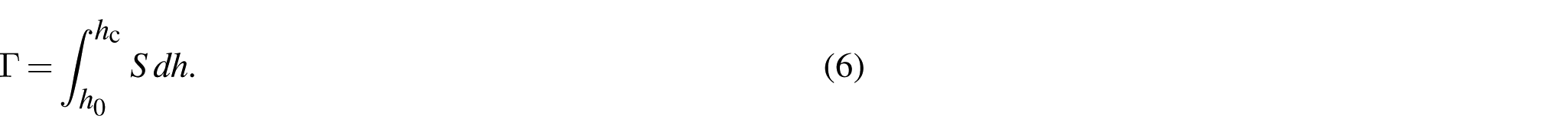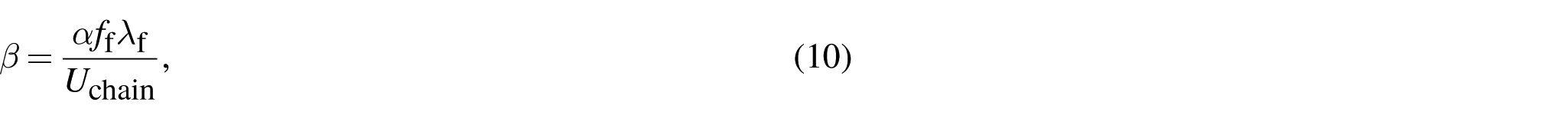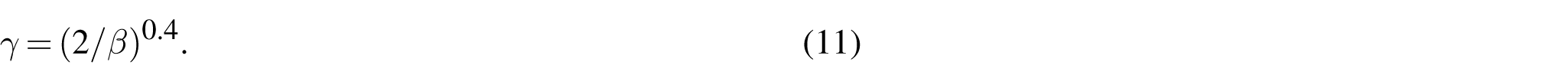The failure resistance of polymer networks dictates their utility as material candidates across industries. However, relating the key length scales driving crack growth to molecular mechanisms remains a key bottleneck in predicting and designing against fracture. The fractocohesive length—defined in terms of the ratio of fracture energy to the specific work to rupture—of a material correlates with the length scale of energy dissipation and controls fracture resistance. Although the Lake–Thomas model predicts the fractocohesive length of a perfect polymer network to match the undeformed mesh size, real soft materials exhibit values that far exceed this prediction. Here, we report extraordinarily high fractocohesive lengths in polymer-like networks with and without defects. We find that even perfect networks can have fractocohesive lengths orders of magnitude higher than the undeformed mesh size due to highly nonlinear chain behavior giving rise to nonlocal effects during fracture. Introducing defects further increases the fractocohesive length. We identify quantitative relations between nonlinear chain mechanics, defect length, defect density, and fractocohesive length. Overall, strain-stiffening chain behavior, defect density, and defect size independently correlate with larger fractocohesive lengths in polymer-like networks, and their individual effects can be collapsed into a single power law scaling. These outcomes point the way towards improved physics-informed design of soft yet tough polymers and metamaterials.
Elastomers and gels are composed of interconnected networks of polymer chains and form the backbone of many common and advanced materials in the modern world. Their capacity to withstand large, reversible deformations while displaying moduli and fracture properties spanning orders of magnitude make them desirable candidates for applications across the biological, medical, automotive, aerospace, and consumer goods sectors. The fundamental material properties controlling the mechanical failure of soft materials play a pivotal role in determining their value in applications [1–3]. Although theoretical models can accurately predict fracture properties of ductile or brittle materials such as metal and glass [4], existing local or linear frameworks do not fully capture the experimentally observed fracture characteristics of real polymer networks [5–9].
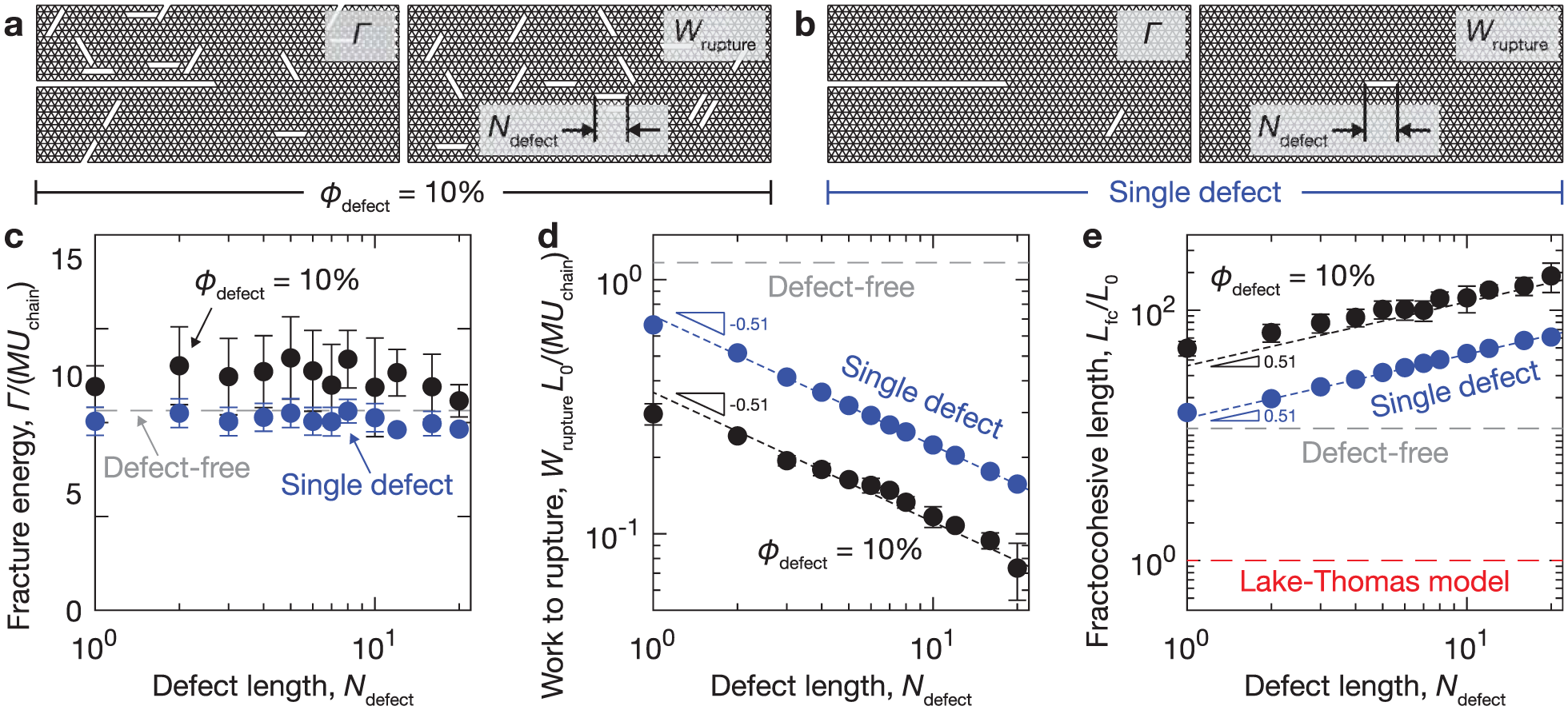
Failure properties of polymer networks are classically measured by two experimental approaches. In the limit of large cracks, the fracture energy Γ describes the energy per unit area (J/m2) required to advance a crack. It is measured by mechanically loading a sample with a single dominant defect until it propagates. In the limit of small cracks, the specific work to rupture describes the energy per unit volume (J/m3) required to break a material. It is measured by uniaxially extending a pristine sample until the material fully separates into multiple parts.
The ratio of the fracture energy and specific work to rupture gives the fractocohesive length scale as [10,11],
Material-specific length scales in fracture like the fractocohesive length have been used experimentally to measure the crack tip diameter [12,13] and characterize flaw sensitivity [11,14,15] in crystalline and amorphous materials. The Lake–Thomas model defines the fracture energy and specific work to rupture in perfect networks, which correspond to network architectures with uniform topologies, monodisperse chain lengths, and no defects. The fracture energy is given as the energy to dissociate a layer of chains as,
where M is the areal density of chains in the undeformed reference state, and is the energy to rupture a single chain. The specific work to rupture is provided as the energy density within the network such that,
where is the undeformed chain length or mesh size. Therefore, the Lake–Thomas model predicts that the fractocohesive length of a perfect network is the original mesh size [16], i.e.,
Real polymers typically exhibit fractocohesive lengths that are orders of magnitude higher than their undeformed mesh sizes, which can reach the centimeter scale. Biological tissues [17] and ultratough hydrogels [18,19] display fractocohesive lengths that may be as large as 1–10 cm. Common polymers like natural rubber [20] and double network gels [21] display fractocohesive lengths of about 0.1 mm. Contributions from sacrificial networks [22–24], chain entanglements [25], hierarchical structures [26,27], strain-induced crystallization [20,28], crosslinking conditions [29], viscoelasticity [30,31], chain dynamics [32], heterogeneities [33], and network imperfection [34] can each enhance the size of the dissipative zone. Understanding the individual molecular and architectural contributions of networks to the fractocohesive length remains a challenging question in the fracture mechanics of stretchable materials.
Here, we implement a polymer-like network model to evaluate computationally the fractocohesive lengths of lattices whose strand mechanical behaviors match those of real flexible polymer chains. We find that perfect polymer-like networks exhibit fractocohesive lengths that are orders of magnitude greater than the undeformed mesh size. The fractocohesive length increases with increasing nonlinearity of the force-extension behavior of polymer chains by promoting nonlocal energy release and dissipation during rupture around the crack tip. Introducing defects further amplifies this effect. Across networks with fixed defect densities, the fractocohesive length increases with defect length, as larger flaws markedly reduce the specific work to rupture. This fracture-modeling approach delivers order-of-magnitude predictive power for the fractocohesive length in polymer-like networks.
2. Methods
Polymer-like networks [35] build on insights from coarse-grained [36–39], mesoscale [40], metamaterial [41], and lattice [42] models by providing an efficient platform to simulate the mechanical response of polymer chains in a crosslinked elastomeric network [43]. Molecular models accounting for entropic elasticity, bond stretching, and chain scission can parametrize experimental single-molecule force spectroscopy (SMFS) measurements of the force-stretch responses from chains of common polymers [44–47]. We adopt the modified freely jointed chain model (m-FJC) to describe the mechanical response of polymer chains within the simulation [45], which relates the force f to the stretch λ through the relationship,
where describes the entropic stiffness, describes the energetic stiffness, and is the transition stretch between these regimes (Figure 1(a)). For polymers, regularizes the freely jointed chain model to describe the effect of bond stretching before chain scission. defines the thermal response as , where is the Boltzmann constant, θ is the absolute temperature, and b is the length of a Kuhn monomer. is the chain stretch at the contour length nb (i.e., ), where n is the number of Kuhn monomers in a polymer chain. Prior works have characterized these values based on SMFS experiments with common polymers [48–53]. Historically, Lake and Thomas considered that the energy required to rupture a chain corresponds to the chemical energy needed to break each of the covalent bonds between the N Kuhn monomers within the polymer chain, i.e., . This view has been challenged by Wang et al. [54] based on force-extension curves obtained through SMFS. Since is not defined in the case of polymer-like networks, we follow Wang et al. [54] and calculate as the integral of the force-stretch response of a polymer chain.
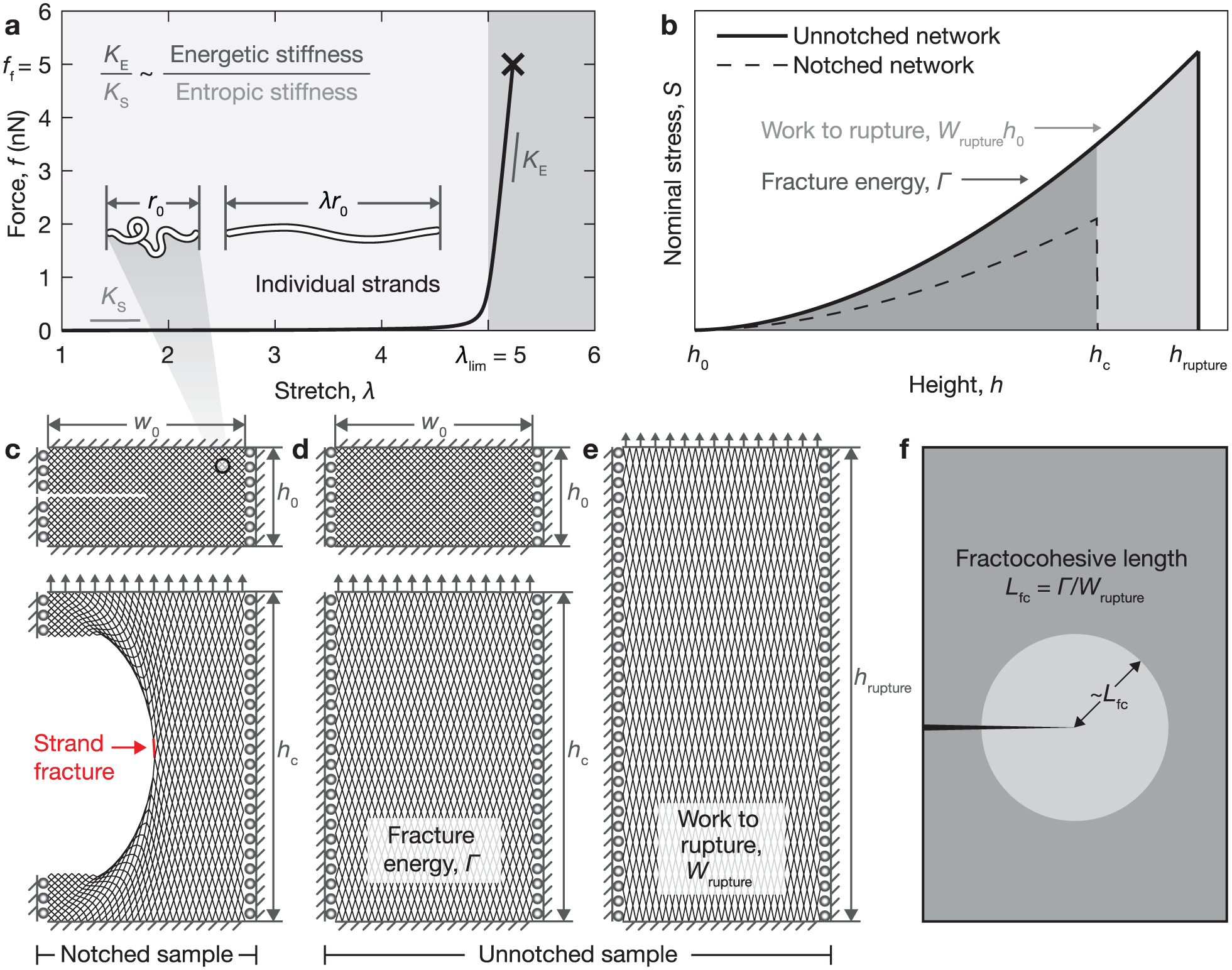
Measuring fractocohesive length in polymer-like networks. (a) Based on single molecule force spectroscopy experiments for a range of flexible polymers [48–53], the nonlinear force-stretch response of polymer chains is modeled by the m-FJC model to transition from entropic unfolding () to bond stretching () at the limiting stretch and break at a fracture force . (b) Fracture energy Γ and specific work to rupture are measured by performing the pure shear fracture test on networks of chains with mechanics governed by the m-FJC model using a quasi-static numerical loading scheme. (c) A notched sample is loaded to the critical height at which the crack propagates by strand fracture. (d) The fracture energy Γ is recorded by loading an unnotched network to . (e) An unnotched sample is loaded to the rupture height at which the network separates. is recorded by integrating this response. (f) The fractocohesive length is the ratio of .
The numerical framework connects strands with behaviors governed by equation (5) into a lattice to perform network-level mechanical simulations. We have provided details of the method in our prior works [35,55,56]; here, we outline all loading schemes applied, parameters selected, and adaptations made. Within a collection of nodes and edges, we store the system energy and minimize it stepwise using Newton’s method while applying boundary conditions across the network. We track the force applied to displace the network F (via the system energy) as a function of sample height h during quasistatic uniaxial extension procedures. The nominal stress is defined as the force divided by the cross-sectional area. This gives in a defect-free two-dimensional (2D) lattice with an undeformed width . The model captures failure by removing chains that reach their rupture criteria; it then recalculates the system state for the new network without that chain connection.
We evaluate Γ and in simulations using two loading schemes (Figure 1(b)). We prescribe to be twice the undeformed height . To measure Γ, we uniaxially extend a notched sample in mode I with an edge crack spanning half of the sample width (). We record the critical height to propagate the edge crack as the sample height at the maximum sample stress (Figure 1(c)). A pristine sample is uniaxially extended from to the rupture height where the lattice separates. We record the nominal stress S during the quasistatic loading process as a function of the sample height h to calculate the energy release rate G. Integrating S from the unnotched sample to (Figure 1(d)) of the notched sample gives
We calculate by integrating the full response of the unnotched sample to (Figure 1(e)) as
In perfect highly stretchable networks, the approximation for as can be applied [57]. Simulations confirm that this result holds for all defect-free polymer-like cases studied here under uniaxial extension with pure shear and tensile loading schemes. Convergence of Γ and with sample size is ensured by simulating networks with up to 1000 layers of strands, as verified by a convergence study [35]. We coarse-grain the architecture away from stress concentrations in homogeneous regions to improve computational efficiency. By removing a few strands that reach their failure criterion to propagate the crack at the critical energy release rate, simulations indicate that the selected specimen geometry and boundary conditions provide adequate constraints to approximately achieve a change in frame between material behind and ahead of the crack tip. We measure by calculating the dividend of from simulations on converged samples (Figure 1(f)). The fractocohesive length scales with the size of the region near the crack tip where the material approaches during fracture. For polymer networks, this measures the extent to which chains approach .
3. Results
We adopt triangular polymer-like networks in two dimensions and measure the fractocohesive length of converged samples while varying the nonlinearity parameter of the m-FJC model to fit the ranges typical of common polymers. SMFS experiments indicate that poly(acrylic acid) (PAA) [48], poly(vinyl alcohol) (PVA) [49], polyisoprene (cis-PI and trans-PI) [50], poly(acryl amide) (PAAM) [51], poly(N-isopropyl acrylamide) (PNIPAM) [51], poly(dimethylacrylamide) (PDMA) [52], poly(diethylacrylamide) (PDEA) [52], and poly(ethylene glycol) (PEG) [53] display ratios of ranging from about to . The transition stretch and rupture force of chains remain constant. The rupture stretch is solved for using equation (5) with a given and . We calculate the fractocohesive length across this range of polymer-like parameters and find that it exceeds the undeformed mesh size in all cases (Figure 2(a)). Values notably elevate with increasing nonlinearity in chain mechanics () up to about 30 times the prediction of the Lake–Thomas model for the case of a network with PEG-like chains ().
Fractocohesive length of polymer-like networks. (a) The measured fractocohesive length is compared with the undeformed mesh size (Lake–Thomas model) with varying ratios of the nonlinearity parameter for common polymers. Experimental single-molecular force spectroscopy measurements give the values of and for poly(acrylic acid) (PAA) [48], poly(vinyl alcohol) (PVA) [49], polyisoprene (cis-PI and trans-PI) [50], poly(acryl amide) (PAAM) [51], poly(N-isopropyl acrylamide) (PNIPAM) [51], poly(dimethylacrylamide) (PDMA) [52], poly(diethylacrylamide) (PDEA) [52], and poly(ethylene glycol) (PEG) [53]. (b) The deformed region near the crack tip of a sample loaded to a stretch of = 2.72 is mapped onto the undeformed configuration for (c-e) to show the released energy in each chain after the bridging strand ruptures (normalized by the breaking energy ). (c) Networks of strands with linear force-stretch relationship (f vs. λ) display fractocohesive lengths near the undeformed mesh size. Networks of strands parametrized to common polymers like (d) PDMA and PDEA and (e) PEG give larger fractocohesive lengths.
We analyze chains surrounding the crack tip to further investigate the relationship between the high fractocohesive lengths and the energetic details associated with chain scission. The polymer-like network model enables an in-depth analysis of failure near the tip of the crack by sequentially providing direct mappings of displacements and strain energy distributions throughout the loading procedure. We save the total stored energy U in each chain as a function of the energy to break the chain at each quasistatic loading step p and apply this to calculate the energy change from steps p to as,
Applying this analysis to the steps before and after scission of the bridging strand gives the released energy after breaking one chain. Visualizations can display the released energy for each chain within a region near the crack tip and map these measurements from the deformed to the undeformed configuration, as depicted in Figure 2(b). We perform a control study on a network of strands with linear force-stretch profiles that break at and . In Figure 2(c), we display the released energy after breaking the bridging strand at the crack tip of the edge-cracked sample from mode I testing. Fracture and rupture tests indicate that is on the same order as the undeformed mesh size in the network of chains with linear mechanics. We highlight this length scale on the undeformed map with a ring surrounding the bridging chain whose radius equals the magnitude of . Chains releasing pronounced energy are concentrated near the crack tip within this network. We repeat this for networks with chains matching the mechanics of PDMA and PDEA in Figure 2(d) and PEG in Figure 2(e). In these polymer-like cases, is larger, and chains releasing significant energy are not concentrated to the few layers surrounding the crack tip. This matches pictures of nonlocal fracture elucidated in computational (discrete, coarse-grained, and continuum) [35,58,59], experimental [5,60,61], and theoretical [6,8,54,62,63] frameworks throughout the literature. The failure zone size near the crack tip—formalized as the dissipative length scale ξ—scales with the fractocohesive length and describes the spatial extent at which the stress and strain concentrations near the tip of the crack decay [10]. Therefore, materials with higher fractocohesive lengths exhibit larger dissipative process zones. We highlight here that the strain-stiffening J-shaped mechanical behavior characteristic of polymer chains with entropic elasticity and bond stretching leads to extraordinarily high values of in polymer-like networks.
Real polymer networks contain defects in the form of dangling ends [6,64], first-order loops [65,66], etc. While network imperfections and inhomogeneities are generally known to increase the value of by orders of magnitude [34], it is difficult to systematically control defects while studying fracture in experiments with real polymers. Similarly, molecular simulations with detailed regulation of defect densities are costly to scale up for bulk fracture simulations containing large cracks. Therefore, due to challenges with conventional measurement techniques, the independent effects of defect size, distribution, and density on the value of remain less understood. We therefore extend the polymer-like simulation platform to model and study networks with defects.
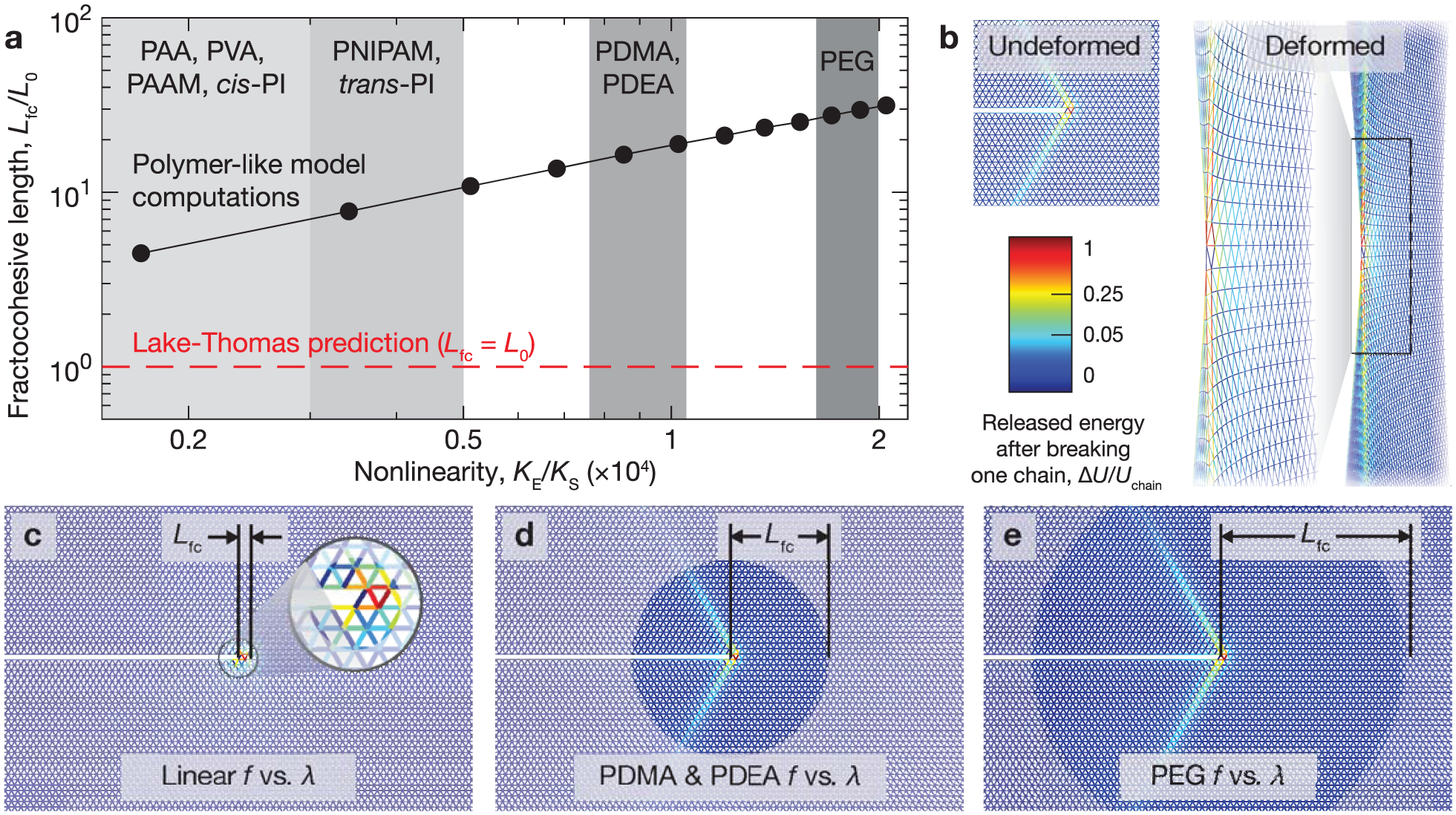
We introduce line defects of length into samples by removing the corresponding edges from the lattice. The values of Γ and can then be calculated using the same approach as is detailed in Figure 1 to give . We first conduct a case study on polymer-like networks with defects of chains with nonlinearity . We fix the defect density to 10% and randomly generate line defects at orientations of 0∘, 60∘, and 120∘ to match the topology of the triangular lattice (Figure 3(a)). We perform a parallel baseline study on networks with a single dominant defect with the goal of decoupling the roles of defect size and density. We randomly orient and place a defect of length in notched samples and place a centered horizontal defect in unnotched samples as illustrated in Figure 3(b). We perform the loading procedures outlined in Figure 1 to measure Γ, , and as a function of defect size in sufficiently large networks without coarse-graining (to avoid error from heterogeneities). Recall that the nominal stress S plotted in Figure 1(b) for loading of pristine samples is defined for perfect 2D networks as . The nominal stress of a sample with a single defect perpendicular to the loading direction can be defined more generally as to account for the decrease in elastically active strands. Note that this approach only marginally affects measurements in all cases measured here since the sample widths are much larger than .
Effect of defect size on the fractocohesive length of polymer-like networks. The fracture energy Γ and specific work to rupture are measured using notched and unnotched polymer-like networks () with (a) a fraction of randomly oriented defects of length and compared with (b) notched and unnotched samples with zero defects or a single dominant defect of length . (c) Γ, (d) , and (e) fractocohesive length (scaled with ) are measured as a function of defect length . The best fits for the and single defect cases give the same scaling exponent with for (d) specific work to rupture () and (e) fractocohesive length (). Error bars depict standard deviations across samples with randomized defect locations and directions.
Calculations depicted in Figure 3(c) indicate that Γ remains approximately constant with respect to defect length at a fixed defect density. We find that this holds for defects whose lengths span less than approximately 10% of the entire sample height used in simulations. Notably, the magnitude of Γ approximately matches the defect-free value. This result holds for networks with and those with a single defect. In contrast, increasing the defect length with a fixed defect concentration decreases (Figure 3(d)). While the magnitude of in the single defect case is higher than the case with 10% defects, the best fit for both cases scale with the same exponent as . The effects of imperfections on the measures of Γ and interact nonlinearly such that increases with defect density and defect length (Figure 3(e)). We fit the fractocohesive length with defect length in the single defect and 10% defects cases and find that they also scale with the same exponent as .
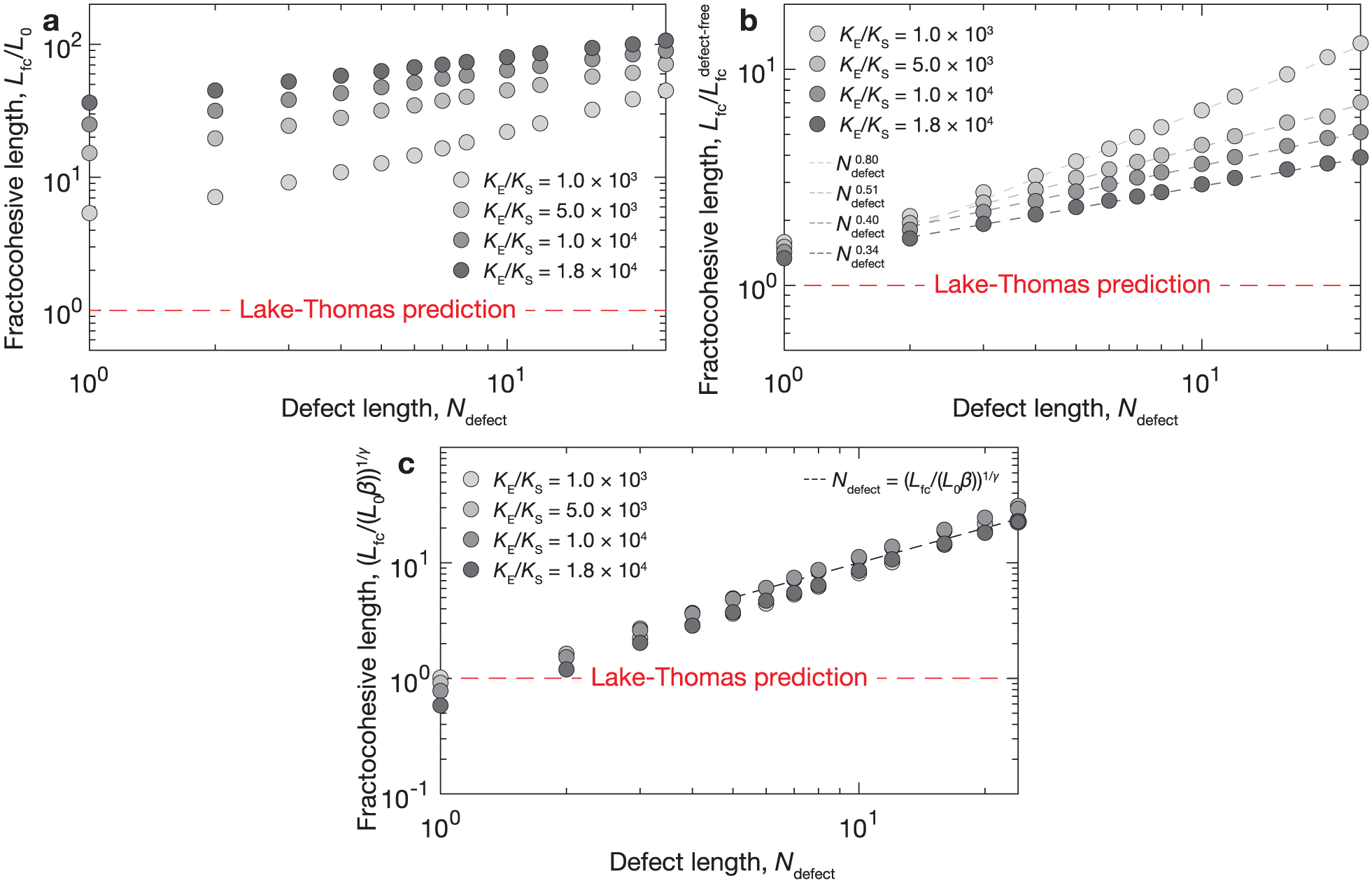
We further investigate the effect of defects in a range of polymer-like networks by tuning the chain nonlinearity parameter . Results from Figure 3 motivate the use of Γ from the defect-free case and from the single defect case to provide a conservative estimate of with respect to the length . We use Γ from the defect-free case because it approximates the magnitude in cases with defects without loss of generality. We use from the single defect case because it gives the same scaling with as the case with random defects. Overall, this strategy results in the same scaling for with as the case with random defects. We apply this approximation technique to measure as a function of defect length while varying . Computations indicate that the fractocohesive length can exceed the undeformed mesh size by over two orders of magnitude in polymer-like networks with defects (Figure 4(a)). We normalize this result by the fractocohesive length of the defect-free sample with the same in Figure 4(b). We find that the power-law scaling with decreases as the nonlinearity parameter increases from for to for .
Scaling for fractocohesive length in polymer-like networks with defects. (a) The variation in fractocohesive length with respect to defect length for varying nonlinearity ratios is conservatively approximated by the defect-free fracture energy and the specific work to rupture of a sample with one dominant defect since the ordering of these values capture the general order of magnitude and ordering of . (b) The scaling of with when normalized by the defect-free case . The legend displays the best fit power law scaling for a range of values of . (c) The outcome of normalizing by the proposed scaling formalized in equation (9) and plotting results with respect to for The scaling relationship predicts that results will collapse to a linear trend on double logarithmic axes (dashed black line).
We now propose a scaling relationship based on these conclusions that predicts the fractocohesive length of perfect and imperfect polymer-like networks. By analyzing how the fractocohesive length scales with each parameter from calculations, the model attempts to resolve contributions from strand mechanics and defects of various sizes. Results indicate that strand nonlinearity and defect length each affect the fractocohesive length in polymer-like systems. We assume the case of a single defect and find that fractocohesive length relates to the undeformed mesh size as,
where the parameter β describes the effect of strain-stiffening in chains and the power law exponent γ describes the scaling with defect length. Note from Figure 4(b) that the scaling with γ depends on and, therefore, is coupled with the nonlinear mechanics of the chain deformation. We can determine relationships for β and γ based on the numerical results. We find that β can be written as,
where α is a geometric parameter describing the lattice topology in 2D and three-dimensional (3D) networks ( for a 2D triangular lattice) that relates to loop size [55]. Note that β directly encodes the effects of strain-stiffening; β increases with in networks of polymer-like chains (see Figure 4a). We find that γ can be written as a function of β by fitting results, giving,
for the triangular lattice topology tested here. Note that equation (9) applies for networks containing defects of a finite size; the defect-free case should instead be modeled using . We normalize the fractocohesive length by the scaling framework presented in equation (9) and plot simulation results against defect length in Figure 4(c) to demonstrate the applicability of the model to networks with . We suspect that geometric effects can account for deviations from the model in predicting results for networks with . This result highlights how implementing the J-shaped force-extension curve of equation (5) that is typical of real polymer chains elevates the fractocohesive length well beyond the undeformed mesh size in perfect networks. Imperfections serve to further increase this ratio. We capture the effect of defect length through the exponent γ. The proposed model provides insights into the roles of polymer-like chains and defects in amplifying length scales involved in soft network fracture.
4. Conclusion
This work raises several open and ongoing questions regarding fracture of polymer and polymer-like networks. For example, subsequent work must be done to fully interpret the physics that underpins the scaling results determined computationally here. Other topological and architectural details that are common in real polymer networks also warrant further analysis, such as chain entanglements [25], interpenetrating networks [21], and polydispersity [67]. More complex damage models incorporating additional volumetric, temporal, and diffusive effects relevant to many real polymer networks could help to address these questions. Results from this work could also have implications for improving gradient-enhanced [58,68–70] and cohesive zone [71–73] fracture models incorporating nonlocal effects. Overall, studying material-specific length scales relevant to fracture through polymer-like network models provides an efficient yet robust computational platform to shed light on the fundamental nonlinear chain mechanics that govern these important problems.
In summary, we show that perfect, defect-free polymer-like networks exhibit fractocohesive lengths that far surpass their undeformed mesh sizes, deviating from the prediction of the Lake–Thomas model. We measure this by performing fracture tests in the limit of small and large cracks on pure shear specimens using a computational polymer-like network model to characterize the fractocohesive length with accurately calibrated molecular mechanics and architectures. Calculations demonstrate that the fractocohesive length exceeds the mesh size by an order of magnitude in networks composed of chains with nonlinear force-extension curves matching the performance of common polymers. We find that it increases when the strain-stiffening force-stretch behavior of chains is more pronounced (i.e., with increasing ). In these notched samples, the energy dissipated by chains during crack growth far outweighs the energy consumed in breaking a layer of strands. Chains exhibiting significant energy release disseminate well beyond the tip of the crack. Dynamic simulations indicate that this promotes a larger energy dissipation zone [35]. Adding imperfections through defects of varying density and size further amplifies these results. In particular, for a given defect density, we demonstrate that the fractocohesive length grows as a function of the defect length. Evidence suggests that these findings can be rationalized by augmenting the prediction of the Lake–Thomas model by incorporating the scalings given by equations (9)–(11). We propose a modified model that relates the fractocohesive length of polymer-like networks to the mechanics of chain extension, the topology of the network architecture, and the length of defects at a given defect density. Our study highlights important molecular details, such as the nonlinear chain force-extension curve along with the number and size of defects in the sample, which promote fracture resistance in polymer networks. It also provides a computationally efficient approach to modeling fractocohesive lengths of diverse elastomeric networks across scales. Our findings not only shed light on soft, tough, and stretchable polymer network design principles but also provide direct implications for developing fatigue-resistant biomaterials and metamaterials for use in domains such as healthcare, aerospace, and soft robotics.
Footnotes
Acknowledgements
The authors acknowledge the MIT SuperCloud and Lincoln Laboratory Supercomputing Center for providing HPC resources that have contributed to the research results reported within this paper.
ORCID iDs
Chase M. Hartquist
Xuanhe Zhao
Funding
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: This work was supported in part by the National Institutes of Health (Grants No. 1R01HL153857-01 and No. 1R01HL167947-01), Department of Defense Congressionally Directed Medical Research Programs (Grant No. PR200524P1), and the National Science Foundation (Grant No. 2430106 and No. EFMA-1935291). The authors also acknowledge support fromstartup funds through the University of Florida.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
1.
CretonCCiccottiM. Fracture and adhesion of soft materials: a review. Rep Prog Phys2016; 79(4): 046601.
2.
ZhaoXChenXYukH, et al. Soft materials by design: unconventional polymer networks give extreme properties. Chem Rev2021; 121(8): 4309–4372.
3.
XuCChenYZhaoS, et al. Mechanical regulation of polymer gels. Chem Rev2024; 124(18): 10435–10508.
4.
GriffithAAVI. The phenomena of rupture and flow in solids. Phil Trans R SocLond Ser A1921; 221(582–593): 163–198.
5.
LivneABouchbinderESvetlizkyI, et al. The near-tip fields of fast cracks. Science2010; 327(5971): 1359–1363.
6.
LinSZhaoX. Fracture of polymer networks with diverse topological defects. Phys Rev E2020; 102(5): 052503.
7.
AkagiYGongJPChungUI, et al. Transition between phantom and affine network model observed in polymer gels with controlled network structure. Macromolecules2013; 46(3): 1035–1040.
8.
BarneyCWYeZSacligilI, et al. Fracture of model end-linked networks. Proc Natl Acad Sci2022; 119(7): e2112389119.
9.
WangSBeechHKBowserBH, et al. Mechanism dictates mechanics: a molecular substituent effect in the macroscopic fracture of a covalent polymer network. JACS2021; 143(10): 3714–3718.
10.
LongRHuiCYGongJP, et al. The fracture of highly deformable soft materials: a tale of two length scales. Annu Rev Conden Ma P2021; 12: 71–94.
ThomasA. Rupture of rubber. II. The strain concentration at an incision. J Polym Sci1955; 18(88): 177–188.
13.
VolokhKTrapperP. Fracture toughness from the standpoint of softening hyperelasticity. J Mech Phys Solids2008; 56(7): 2459–2472.
14.
GaoHJiBJägerIL, et al. Materials become insensitive to flaws at nanoscale: lessons from nature. Proc Natl Acad Sci2003; 100(10): 5597–5600.
15.
ZhangTLiXKadkhodaeiS, et al. Flaw insensitive fracture in nanocrystalline graphene. Nano letters2012; 12(9): 4605–4610.
16.
LakeGLindleyP. The mechanical fatigue limit for rubber. J Appl Polym Sci1965; 9(4): 1233–1251.
17.
ZengLLiuFYuQ, et al. Flaw-insensitive fatigue resistance of chemically fixed collagenous soft tissues. Sci Adv2023; 9(9): eade7375.
18.
MaJZhangXYinD, et al. Designing ultratough single-network hydrogels with centimeter-scale fractocohesive lengths via inelastic crack blunting. Adv Mater2024; 36(23): 2311795.
19.
HanZWangPLuY, et al. A versatile hydrogel network–repairing strategy achieved by the covalent-like hydrogen bond interaction. Sci Adv2022; 8(8): eabl5066.
20.
ThomasA. Rupture of rubber. V. Cut growth in natural rubber vulcanizates. J Polym Sci1958; 31(123): 467–480.
21.
GongJPKatsuyamaYKurokawaT, et al. Double-network hydrogels with extremely high mechanical strength. Adv Mater2003; 15(14): 1155–1158.
22.
HaqueMAKurokawaTKamitaG, et al. Lamellar bilayers as reversible sacrificial bonds to toughen hydrogel: hysteresis, self-recovery, fatigue resistance, and crack blunting. Macromolecules2011; 44(22): 8916–8924.
23.
SunJYZhaoXIlleperumaWR, et al. Highly stretchable and tough hydrogels. Nature2012; 489(7414): 133–136.
24.
DucrotEChenYBultersM, et al. Toughening elastomers with sacrificial bonds and watching them break. Science2014; 344(6180): 186–189.
25.
KimJZhangGShiM, et al. Fracture, fatigue, and friction of polymers in which entanglements greatly outnumber cross-links. Science2021; 374(6564): 212–216.
26.
WuXLiXSunS, et al. Fracture process zone and fracture energy of heterogeneous soft materials. J Mech Phys Solids2025; 196: 105997.
27.
ChenQChenBJingS, et al. Flaw sensitivity of cellulose paper. Extreme Mech Lett2022; 56: 101865.
28.
HartquistCMLinSZhangJH, et al. An elastomer with ultrahigh strain-induced crystallization. Sci Adv2023; 9(50): eadj0411.
29.
LiCWangZWangY, et al. Effects of network structures on the fracture of hydrogel. Extreme Mech Lett2021; 49: 101495.
DengBWangSHartquistC, et al. Nonlocal intrinsic fracture energy of polymerlike networks. Phys Rev Lett2023; 131(22): 228102.
36.
LeiJLiZXuS, et al. A mesoscopic network mechanics method to reproduce the large deformation and fracture process of cross-linked elastomers. J Mech Phys Solids2021; 156: 104599.
37.
GhareebAElbannaA. An adaptive quasicontinuum approach for modeling fracture in networked materials: application to modeling of polymer networks. J Mech Phys Solids2020; 137: 103819.
38.
LeiJLiuZ. A network mechanics method to study the mechanism of the large-deformation fracture of elastomers. J Appl Phys2022; 132(13): 135101.
39.
AroraA. Effect of spatial heterogeneity on the elasticity and fracture of polymer networks. Macromolecules2025; 58(2): 1143–1155.
YamaguchiTOnoueYSawaeY. Topology and toughening of sparse elastic networks. Phys Rev Lett2020; 124(6): 068002.
42.
PanZMaRWangD, et al. A review of lattice type model in fracture mechanics: theory, applications, and perspectives. Eng Fract Mech2018; 190: 382–409.
43.
BroederszCPMacKintoshFC. Modeling semiflexible polymer networks. Rev Mod Phys2014; 86(3): 995.
44.
KuhnW. Dependence of the average transversal on the longitudinal dimensions of statistical coils formed by chain molecules. J Polym Sci1946; 1(5): 380–388.
45.
SmithSBCuiYBustamanteC. Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science1996; 271(5250): 795–799.
46.
MaoYAnandL. A theory for fracture of polymeric gels. J Mech Phys Solids2018; 115: 30–53.
LiHLiuBZhangX, et al. Single-molecule force spectroscopy on poly (acrylic acid) by AFM. Langmuir1999; 15(6): 2120–2124.
49.
LiHZhangWZhangX, et al. Single molecule force spectroscopy on poly (vinyl alcohol) by atomic force microscopy. Macromol Rapid Comm1998; 19(12): 609–611.
50.
ZhangW. Nano-mechanical detection of single molecules. PhD Thesis, Jilin University, Changchun, China, 2002.
51.
ZhangWZouSWangC, et al. Single polymer chain elongation of poly (n-isopropylacrylamide) and poly (acrylamide) by atomic force microscopy. J Phys Chem B2000; 104(44): 10258–10264.
52.
WangCShiWZhangW, et al. Force spectroscopy study on poly (acrylamide) derivatives: effects of substitutes and buffers on single-chain elasticity. Nano Lett2002; 2(10): 1169–1172.
53.
OesterheltFRiefMGaubH. Single molecule force spectroscopy by AFM indicates helical structure of poly (ethylene-glycol) in water. New J Phys1999; 1(1): 6.
54.
WangSPanyukovSRubinsteinM, et al. Quantitative adjustment to the molecular energy parameter in the Lake–Thomas theory of polymer fracture energy. Macromolecules2019; 52(7): 2772–2777.
55.
HartquistCWangSCuiQ, et al. Scaling law for intrinsic fracture energy of diverse stretchable networks. Phys Rev X2025; 15(1): 011002.
56.
HartquistCMWangSDengB, et al. Fracture of polymer-like networks with hybrid bond strengths. J Mech Phys Solids2025; 195: 105931.
57.
OrowanE. Fracture and strength of solids. Rep Prog Phys1949; 12(1): 185.
58.
TalaminiBMaoYAnandL. Progressive damage and rupture in polymers. J Mech Phys Solids2018; 111: 434–457.
59.
MousaviSMMulderrigJTalaminiB, et al. A chain stretch-based gradient-enhanced model for damage and fracture in elastomers. Comput Method Appl Mech Eng2025; 444: 118103.
60.
XueNLongRDufresneER, et al. Elastomers fail from the edge. Phys Rev X2024; 14(1): 011054.
61.
LuWWangCZhouZ, et al. Quantify the fracture process zone and elastic dissipation zone: a new insight into intrinsic fracture of polymer networks. Extreme Mech Lett2025; 78: 102362.
62.
WangSPanyukovSCraigSL, et al. Contribution of unbroken strands to the fracture of polymer networks. Macromolecules2023; 56(6): 2309–2318.
63.
WangSHartquistCMDengB, et al. A loop-opening model for the intrinsic fracture energy of polymer networks. Macromolecules2024; 57(13): 6069–6075.
64.
Di LorenzoFSeiffertS. Nanostructural heterogeneity in polymer networks and gels. Polym Chem2015; 6(31): 5515–5528.
65.
ZhouHWooJCokAM, et al. Counting primary loops in polymer gels. Proc Natl Acad Sci2012; 109(47): 19119–19124.
66.
PanyukovS. Loops in polymer networks. Macromolecules2019; 52(11): 4145–4153.
67.
AroraALinTSBeechHK, et al. Fracture of polymer networks containing topological defects. Macromolecules2020; 53(17): 7346–7355.
VernereyFJBrighentiRLongR, et al. Statistical damage mechanics of polymer networks. Macromolecules2018; 51(17): 6609–6622.
70.
MulderrigJLiBBouklasN. Affine and non-affine microsphere models for chain scission in polydisperse elastomer networks. Mech Mater2021; 160: 103857.
71.
ElicesMGuineaGGomezJ, et al. The cohesive zone model: advantages, limitations and challenges. Eng Fract Mech2002; 69(2): 137–163.
72.
ParkKPaulinoGH. Cohesive zone models: a critical review of traction-separation relationships across fracture surfaces. Appl Mech Rev2011; 64(6): 060802.
73.
HuiCRuinaALongR, et al. Cohesive zone models and fracture. J Adhes2011; 87(1): 1–52.