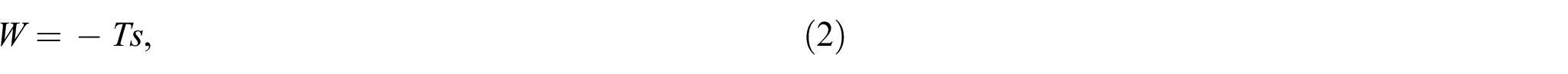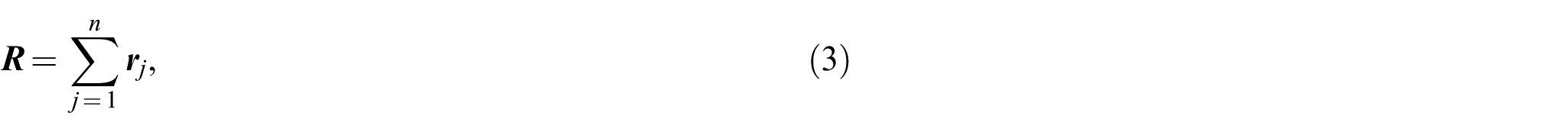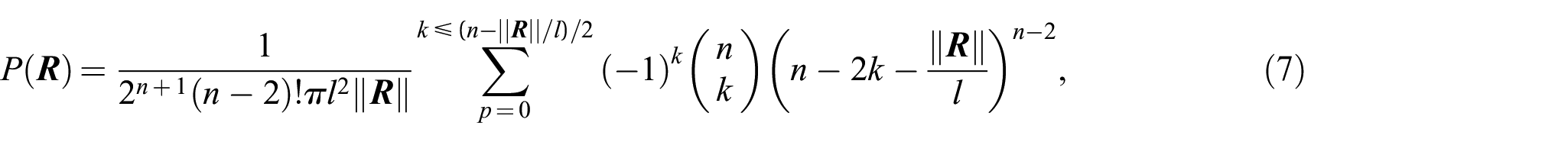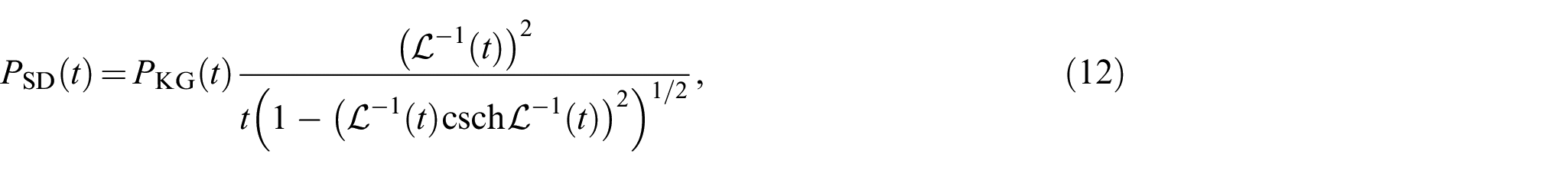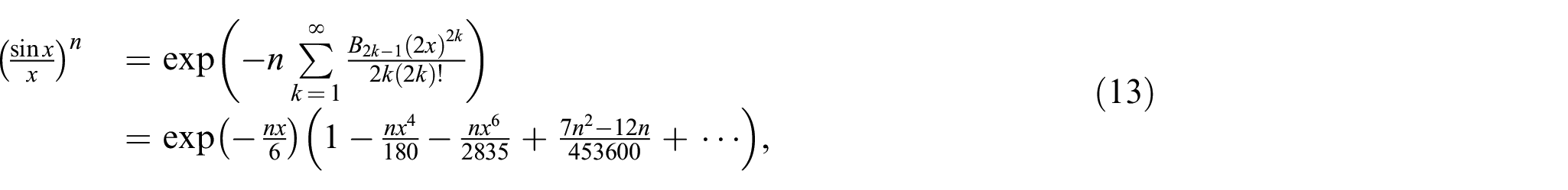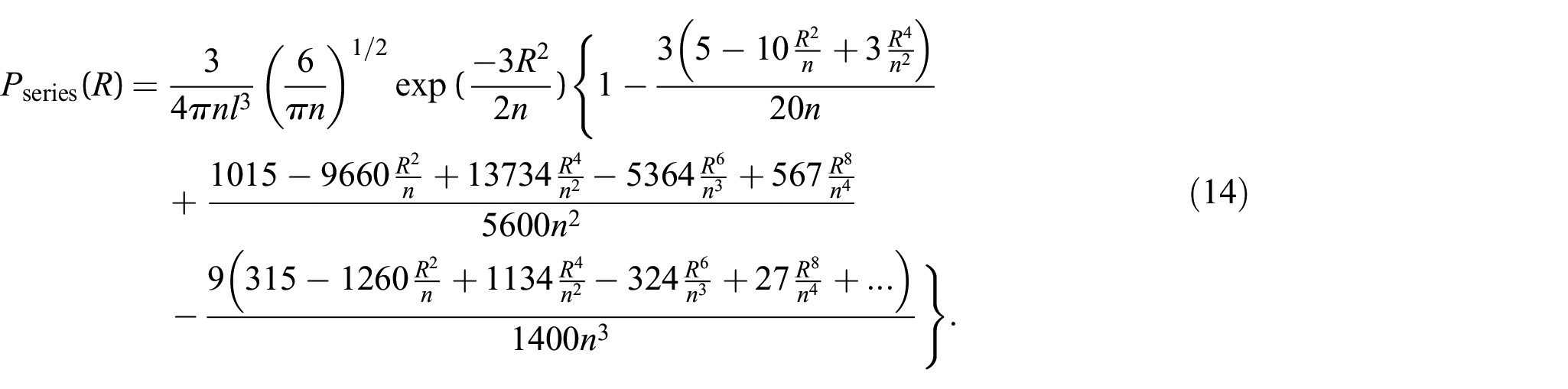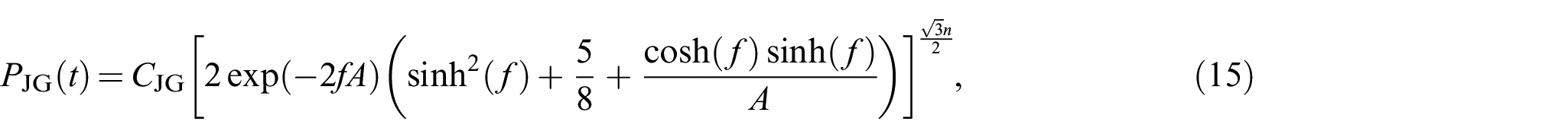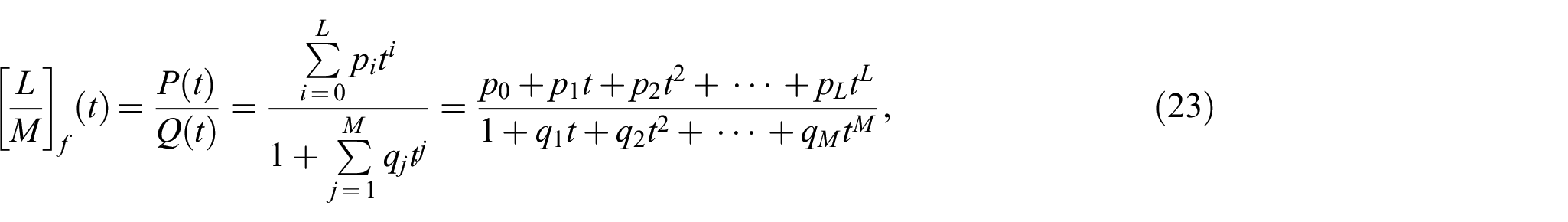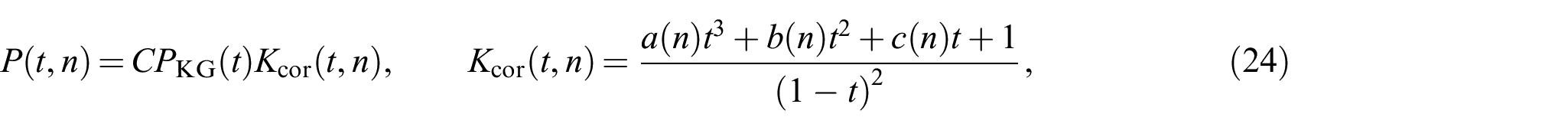Network models of rubber elasticity are based on the conformational entropy of an idealized chain and mostly motivated by the non-Gaussian statistical theory by Kuhn and Grün. However, the non-Gaussian probability distribution function cannot be expressed in a closed form and requires an approximation. All such approximations applied in the literature demonstrate pronounced inaccuracies in comparison to the analytical solution. The ideal choice of the approximation function depends on a variety of factors, such as the chain parameters or the desired application of the approximation (the probability distribution function itself, the corresponding entropic energy, or the force developed by the chain). In addition, when making a choice regarding the best approximation for a given application, the applied error measure plays a significant role since the approximation that grants, for example, the best maximal relative error is not necessarily the same that provides the best mean absolute error. In the literature, this application-specific evaluation of available approximations is commonly disregarded. In this paper, we evaluate previously proposed approximations on the application-specific basis and develop an approach to derive a family of approximations for the free energy of a polymer chain in a broader range of the number of its chain segments. The analytical method based on the Padé technique delivers an approximation of the non-Gaussian probability distribution function that can be easily tailored depending on the desired application. The proposed approach is capable to provide much stronger predictions in comparison to the Kuhn and Grün model in a wide range of chain segment numbers.
A statistical theory of a single long-chain polymer molecule is an essential basis for developing a micromechanical model for rubbers. Traditional molecular simulations take into account all chemical aspects at the molecular level, and are therefore very powerful and comprehensive [1]. However, this detailed information is essential only in microscopic dimensions or small-time ranges of up to 1 ms. Due to the computational expenses and limitations to size and calculation time of such simulations, statistical averaging to larger scales is of interest. The majority of the rubber elasticity network models are based on the statistical theory of an idealized chain and in particular on the freely jointed chain (FJC) model. It is the most simplified representation of a real polymer molecule that can randomly fluctuate in space.
In polymer physics, the probability distribution function (PDF) of an FJC with fixed end positions can be described by the random walk theory. The random walk theory describes a random movement of a particle where a direction in each step is independent of all previous steps. If the length of each step is constant, the particle trajectory is one of all possible conformations of the FJC model. The conformational entropy of the chain is directly related to the PDF and, thus, to the end-to-end distance by the Boltzmann equation
where denotes the Boltzmann constant. The free energy of an ideal polymer chain is derived from its conformational entropy, as
where denotes the absolute temperature. Thus, from a point of view of constitutive modeling, finding an approximation for that leads to a minimized error in is of larger interest than minimizing the approximation error of itself.
The first application of the random walk theory to polymer chain statistics was independently proposed by Kuhn [2] and Guth [3]. Kuhn and Grün [4] introduced the inverse Langevin approximation for single-chain statistics for large deformations which appears accurate for relatively long polymer chains. Many constitutive models of rubber elasticity incorporated the Kuhn–Grün approximations of the non-Gaussian theory [5–8]. These are, for example, the three-chain [9], the four-chain [10], the eight-chain [5], the micro-macro unit sphere model [7], and full-network models [8,11,12]. However, deviations of the Kuhn–Grün model from the exact distribution function in the cases of large extensibility and short chains have motivated to search for more accurate alternatives [9,13,14]. Moreover, the inverse Langevin function cannot be represented in an explicit form and requires an approximation, which increases the deviation from the exact distribution function.
As a result, all models proposed so far suffer from the inaccuracies in the Kuhn–Grün approximation. While some approximations outperform the others in the majority of cases, there is no explicitly superior approximation. The ideal choice of the approximation function depends on a variety of factors, such as the chain parameters, or the desired application of the approximation (the probability distribution function itself, the corresponding free energy, or the entropic force developed by the chain). In addition, when making a choice regarding the best approximation for a given application, the applied error measure plays a significant role since the approximation that grants, for example, the best maximal relative error is not necessarily the same that provides the best mean absolute error.
It is hence reasonable to choose an approximation that suits the error measure deemed most critical for the desired application. In this regard, it appears questionable to take one specific approximation as the final solution that fits all conditions. All previously proposed approximations perform well only for a particular number of chain segments while the effect of remains undiscussed. In this paper, we develop an approach to derive a family of approximations for the free energy of a polymer chain with a broader range of chain segments.
2. Statistics of a single chain
Chain-like molecules such as a single polymer chain in a dilute polymer solution consist of relatively small repeating units (monomers). They are covalently bonded together at some position referred to as cross-linking junctions. In an equilibrium state, the polymer chain can take a definite number of configurations between two end points.
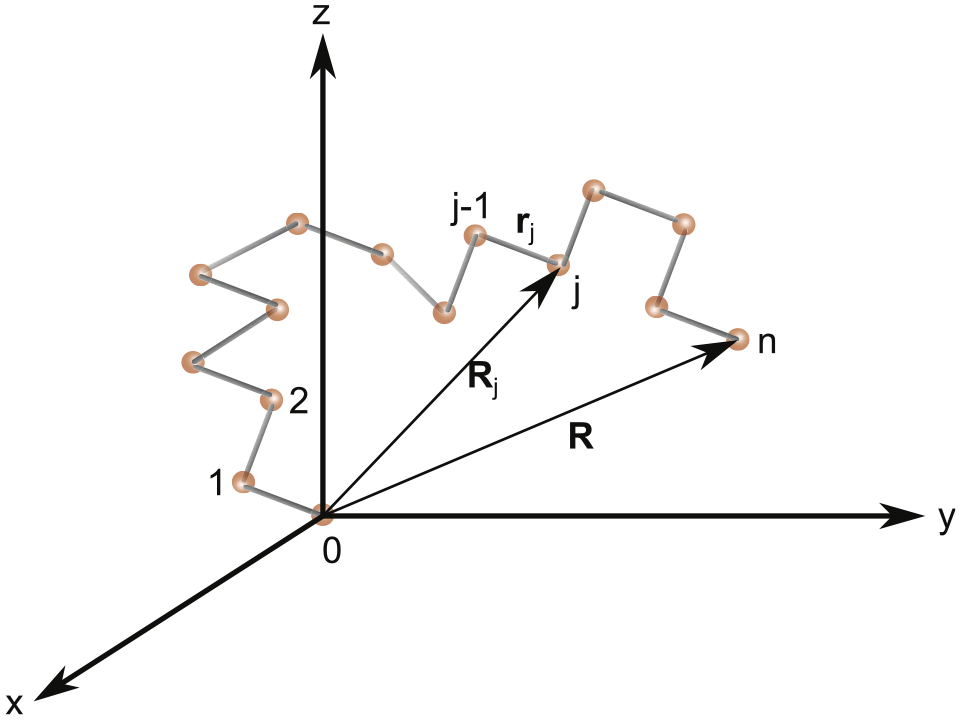
By ignoring the variation of the length of a valence bond from its mean value due to atomic vibrations in the chain molecules [15], one can assume that each bond has a constant length . The distribution function of the end-to-end distance vector subject to the following condition (see Figure 1)
Configuration of a polymer chain with segments and the first segment placed at the origin of a coordinate system.
If the mean end-to-end distance is much less than the maximum extension of the chain , equation (4) reduces to the Gaussian distribution function
Most of the problems in polymer physics are treated based on the Gaussian chain model (5), which is physically unreasonable for real chains [15] in particular in the case of large deformations.
The probability distribution function is subject to the normalization condition generally written by [17]
Equation (4) is given in the integral form and is thus not convenient for practical applications and numerical computation, particularly for large values of . Rayleigh [18] obtained a solution for small values of , e.g., . Chandrasekhar [19] evaluated the integral equation (4) for any finite value of separately. Nevertheless, the calculations become very tedious for larger values of . There have been several attempts to get an exact solution for equation (4). Treloar [20], Wang and Guth [9], Nagai [21], and Hsiung et al. [22] represented it in form of a series as
which is also inconvenient for practical calculation, especially for large values of . For this reason, it is usually approximated by a closed-form expression. In the following section, the most well-known approximations of the exact probability distribution function are discussed and compared with respect to their accuracy.
3. Appraisal and comparison of different approximations
In order to avoid tedious and computationally expensive calculation of the integral form (4) or its series expansion (7), several approximations based on different methods and approaches have been proposed. It is clear that a robust and comprehensive approximation must be capable of fitting to the exact solution for all ranges of the variables , , and or at least to their physical ranges for a specific application. In order to verify the effects of these parameters, we introduce an end-to-end extension ratio such that , and where the abbreviation is used. In the following, different methods approximating the exact solution are discussed and compared.
3.1. Asymptotic analysis
Based on the asymptotic analysis [23], Kuhn and Grün [4], James and Guth [24], and Flory and Rehner [10] obtained an approximation for equation (4) for the case and . Their results are the same but follow distinctive approaches resulting from the non-Gaussian distribution with limited chain extension. The Kuhn–Grün approximation is given by
where represents the inverse of the Langevin function defined as . An accurate approximation of this function can be based on a high-order series expansion (see, e.g., Itskov et al. [25]), on a non-rational or a rational function. Recently, a number of approximations have been proposed for it (26–28). In the limit of , equation (8) results in the Gaussian distribution (5).
An amended Kuhn–Grün approximation proposed by Jernigan and Flory [14] aims to describe polymeric network chains in the context of rubber elasticity at high extensions as
where denotes the normalization factor satisfying equation (6).
3.2. Steepest descent method
The steepest descent or saddle-point method is an extension of Laplace’s method for an integral approximation. By the steepest decent, Wang and Guth [9] obtained the solution of equation (4) for as
with , where and are given by
In a similar way, Yamakawa [16] also derived the following expression
where csch represents the cosecans hyperbolicus. It should be noted that equation (12) is valid over the whole range of .
3.3. Series expansions
Most of the proposed approximations focus mainly on the simplified and specific solutions of the exact problem like series for the inverse of Langevin function. Most of the Taylor series expansions are developed around and are only valid for limited extensions of the polymer chain. In case of (very long chains), the Taylor series for is written as
Other approximations proposed in the literature are either the modified and enhanced version of the aforementioned approximations or formulated based on a combination of the previous approaches. For a model corresponding to a random walk on a diamond lattice, James and Guth [24] obtained an expression (unpublished but mentioned by Wang and Guth [9]) for the case of perfectly flexible chains as
where and must be extracted from
Malacarne et al. [29] proposed an approximation based on the non-Gaussian Tsallis distribution which is an improved version of the Gaussian model. Their proposal is efficient and simple to implement for a large number of monomers. Accordingly, the normalized distribution takes the form as
where represents the gamma function.
Ilg et al. [13] proposed an estimation based on a one-dimensional version of a finitely extensible dumbbell model. The proposal utilizes trigonometric functions to approximate the inverse Langevin function in the formula by Kuhn and Grün [4] as
where is the normalization factor. Recently, Khiêm and Itskov [30] revised the approximation by Ilg, equation (18), as
where denotes the normalization factor.
Very recently, Morovati and Dargazany [31] proposed multiplicative corrections of the Kuhn–Grün approximation (8) as
Note that all proposed approximation functions must satisfy the normalization condition (6).
4. Comparison and discussion of existing approximations
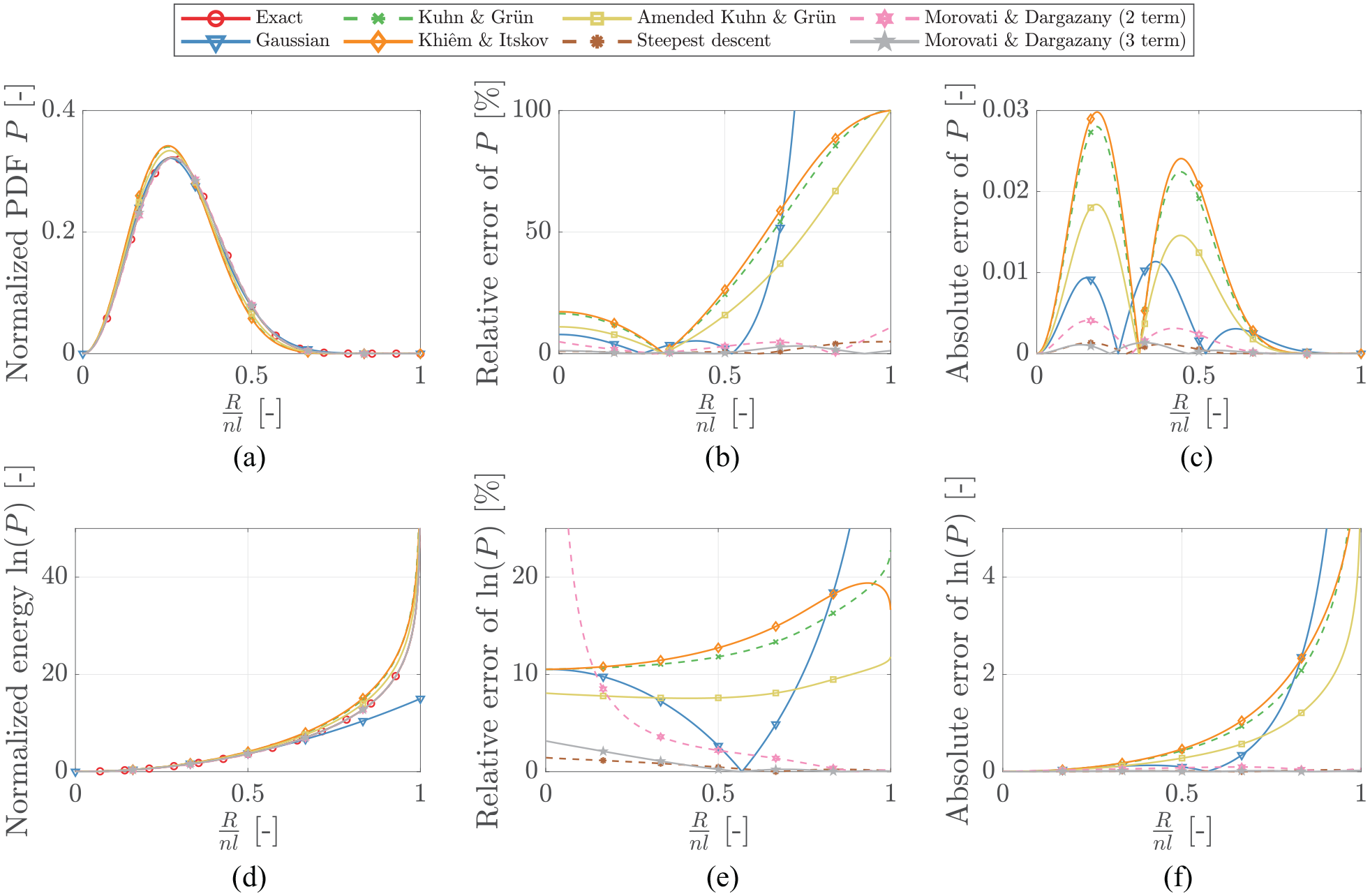
The above discussed approximations are compared in Figure 2 for the case of . Both the normalized PDFs and the corresponding normalized entropic energies are shown along with their respective relative and absolute errors. Generally, relative errors tend to provide a better assessment of the approximative performance. However, this is not the case for the entire range of as the exact PDF and all its approximations converge to zero with approaching one. This leads to extremely small values and large relative errors. In cases like these, the relative error is not meaningful. Hence, it is reasonable to judge the approximative capabilities by utilizing the absolute and relative errors side-by-side.
Comparison of the different normalized PDFs and the corresponding normalized energies along with their respective relative and absolute errors.
Accordingly, the steepest descent, equation (12), and the three-term Morovati and Dargazany approximation, equation (21), significantly outperform the others for the example case of . However, this conclusion cannot be generalized for arbitrary without further investigation. All previously proposed approximations have been proven to perform well in single cases chosen by the respective authors leaving other important domains of undiscussed. Thus, a deeper investigation of the approximations over a reasonable range for is necessary to ensure proper comparability.
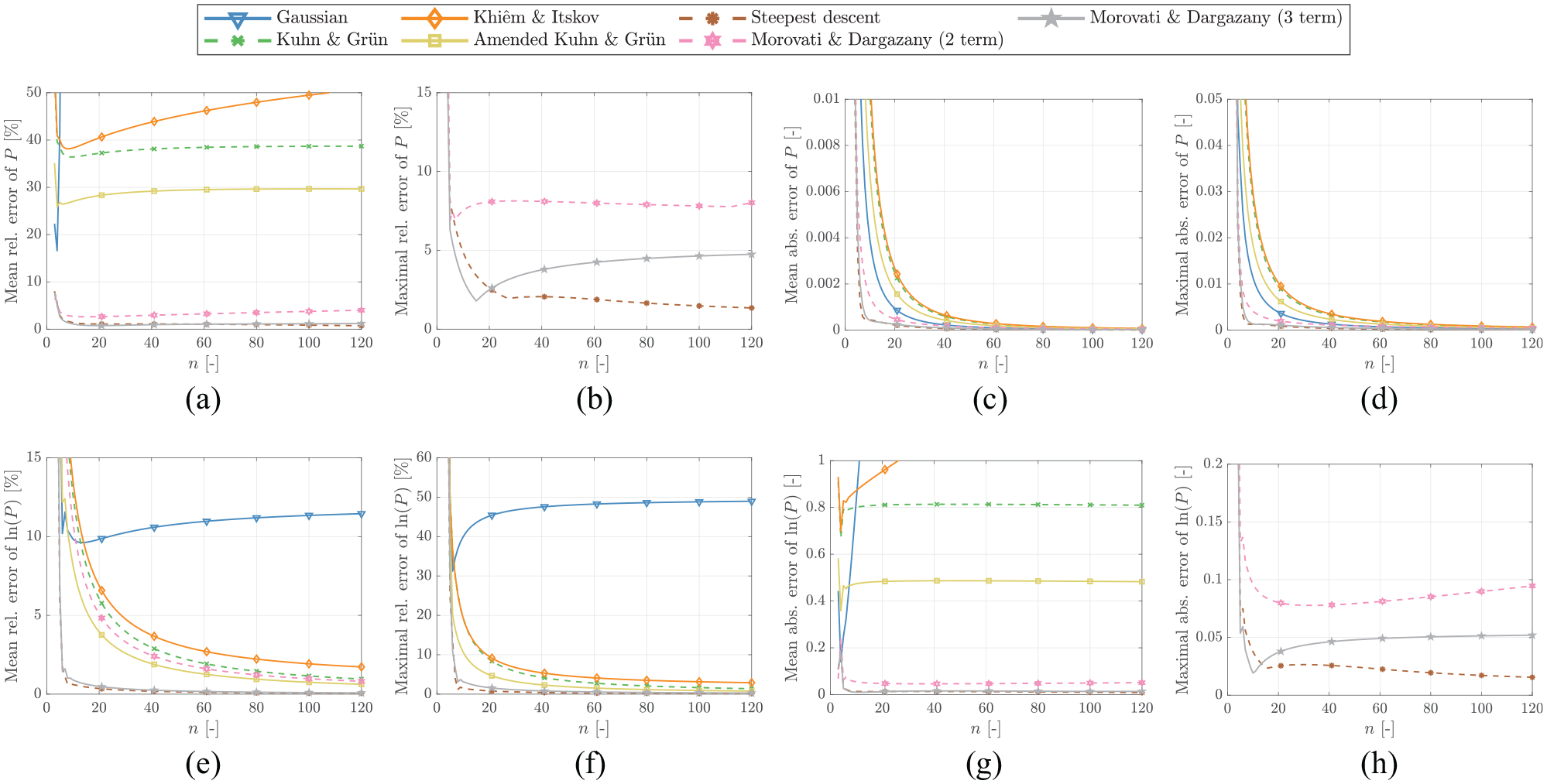
In the following, all approximations discussed in the previous sections are evaluated for every integer within . For each case, the absolute and relative errors were computed. The mean and the maximum of these errors are plotted in Figure 3. Extreme extensibility is less important for practical applications as the chain force tends to infinity. Therefore, in this study, the errors are only evaluated in a range limited to . This limitation allows the comparison of different models in a more practically relevant and realistic way.
Comparison of the mean and average errors of different normalized PDFs (top row) and normalized energies (bottom row) over a range of . Note that some curves with large errors have been removed while cropping the subfigure to the area of interest.
According to the results plotted in Figure 3, the approximative qualities of the approaches vary significantly for different . While there are some approximations that outperform the others in the majority of cases, there is no clear superior approximation in the whole domain and with respect to every considered error measure. In order to find the most suitable approximation, one hence has to base the choice on the specific value of of the polymer chain studied, whether the PDF itself or the corresponding entropic energy should be approximated, and the error measure deemed most important for the considered application. Since the exact solution (7) explicitly depends on the choice of , a suitable approximation can be expected to do as well. However, all previously discussed approximations lack this feature.
5. An improved approximation based on a Padé approximant
Many previously discussed approximations share a common structure containing of up to three parts. The core of these approximations is the Kuhn–Grün approximation corrected by a factor . Finally, the normalization condition (6) is enforced using a scaling factor , which results in a generalized approximation formula as
In the present work, a correction function based on a Padé approximation is proposed. For a given function , the Padé [32–34] approximant of order [L/M] is a rational function of the form
where and denote the integers determining the order of the polynomials in the nominator and denominator, respectively. The choice of does not limit the generality but provides uniqueness of the Padé approximant for given and .
In the present work, we set and , and consider the maximal extensibility of a polymer chain at (where ). Accordingly, a PDF based on the Padé approximation can be given by
with three parameters , , and each depending on . can be calculated in a straight-forward fashion using the normalization condition (6). For convenience, can also be factored into , which leads to and linearly scales , , and . Practically, this can be achieved by either optimizing for , , and and then calculating equation (6) or directly applying the normalization as boundary condition while optimizing for , , , and . Both procedures will provide the same solution.
Usually, the parameters , , and are obtained from partial sums of the Taylor series expansion around some point [35]. This is not feasible in the present context, where the function to approximate is not given in an explicit form and hence cannot be utilized. For this reason, the Levenberg–Marquardt method [36,37] implemented in MATLAB [38] was applied to obtain the parameters , , and for every particular value of .
The specific target function for the optimization procedure has a special significance discussed in the following. While a least-squares-based approach targeting the PDF itself may be an obvious choice, it is neither the only nor necessarily the best one. Depending on the desired application, minimizing the relative error or the mean/maximum of one of the error norms can be advantageous. For example, it appears reasonable to focus on the optimization of rather than on itself due to the higher significance of the normalized energy compared to the pure PDF in the context of constitutive modeling of polymers.
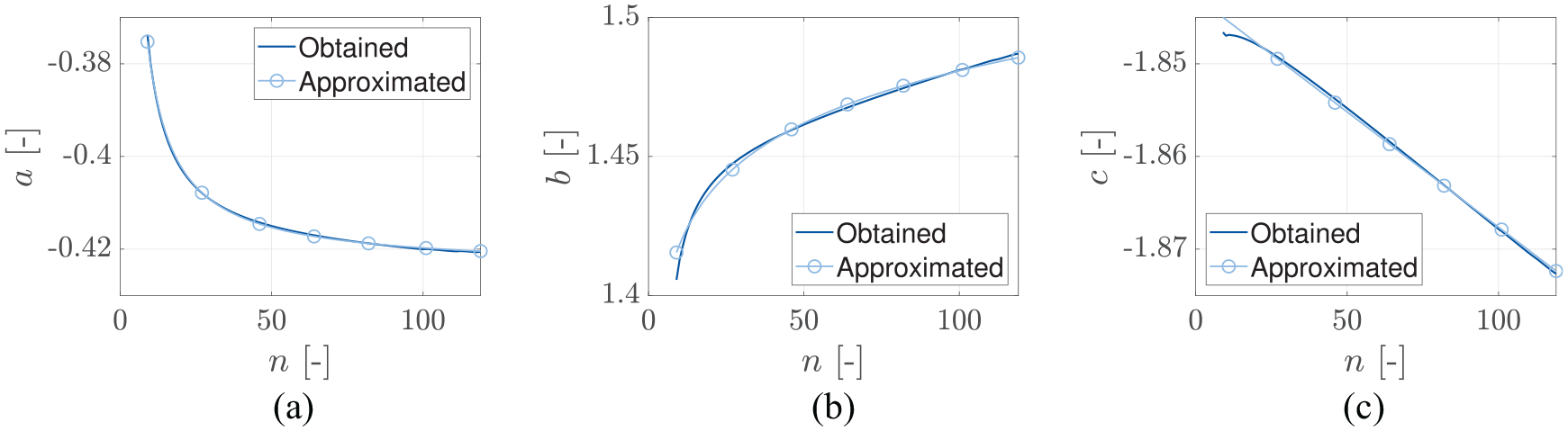
For the sake of this exemplary study, we minimized the least-squares of the absolute error of . The so resulting values for , , and are shown in Figure 4 as functions of . This allows for perfect tailoring of the approximation to a specific application. In the relevant range of , the parameters , , and show a behavior, which can be easily fitted to simple functions, allowing quick and easy calculation (without any new optimization). The corresponding functions, as depicted in Figure 4, can be represented as
Parameters , , and obtained for up to . The optimization target was to minimize the least-squares of the absolute error of . In addition, an approximation for each curve is given.
6. Results
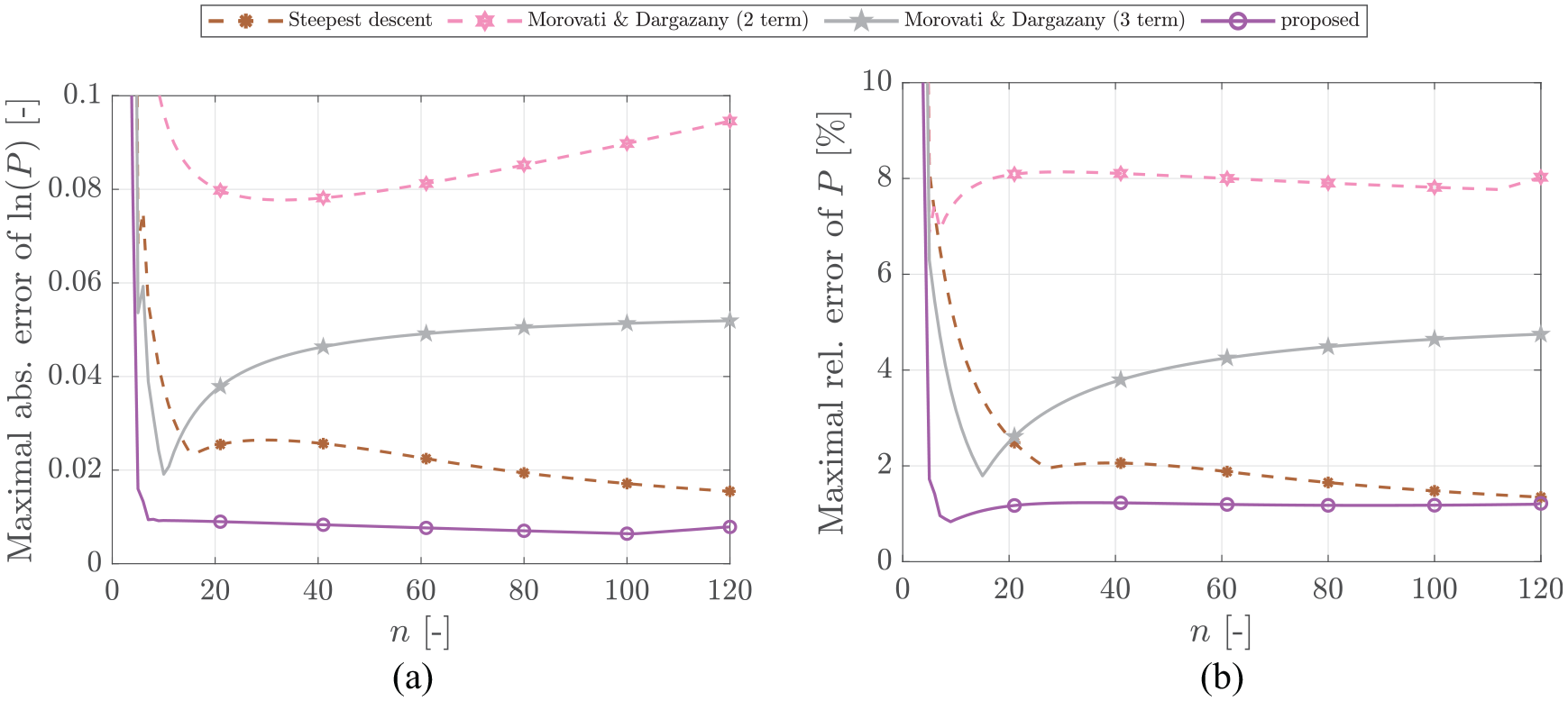
In the following, the Padé approximation-based approach featuring the functional dependence on , as proposed in the previous section, is compared to the steepest descent approach and both the two-term and the three-term Morovati and Dargazany approximation which revealed the best performance in comparison to other approximations in a wide range of .
In Figure 5, the maximal absolute error of and the maximal relative error of are plotted versus . One observes that the proposed approximation demonstrates the best performance in the whole range of . In addition, the proposed approach leads to an error less dependent on . While the advantage is especially prominent for the absolute error of , which was set as the optimization target, it is still present in the PDF itself. Except for very short chains , the relative error of the PDF is never higher than 1.3% and the absolute error of never exceeds .
Comparison of the Padé-based approximation as given in equation (24) to the best approximations from the literature. The left picture shows the absolute error of (which was set as optimization target), while the right shows the resulting relative error of .
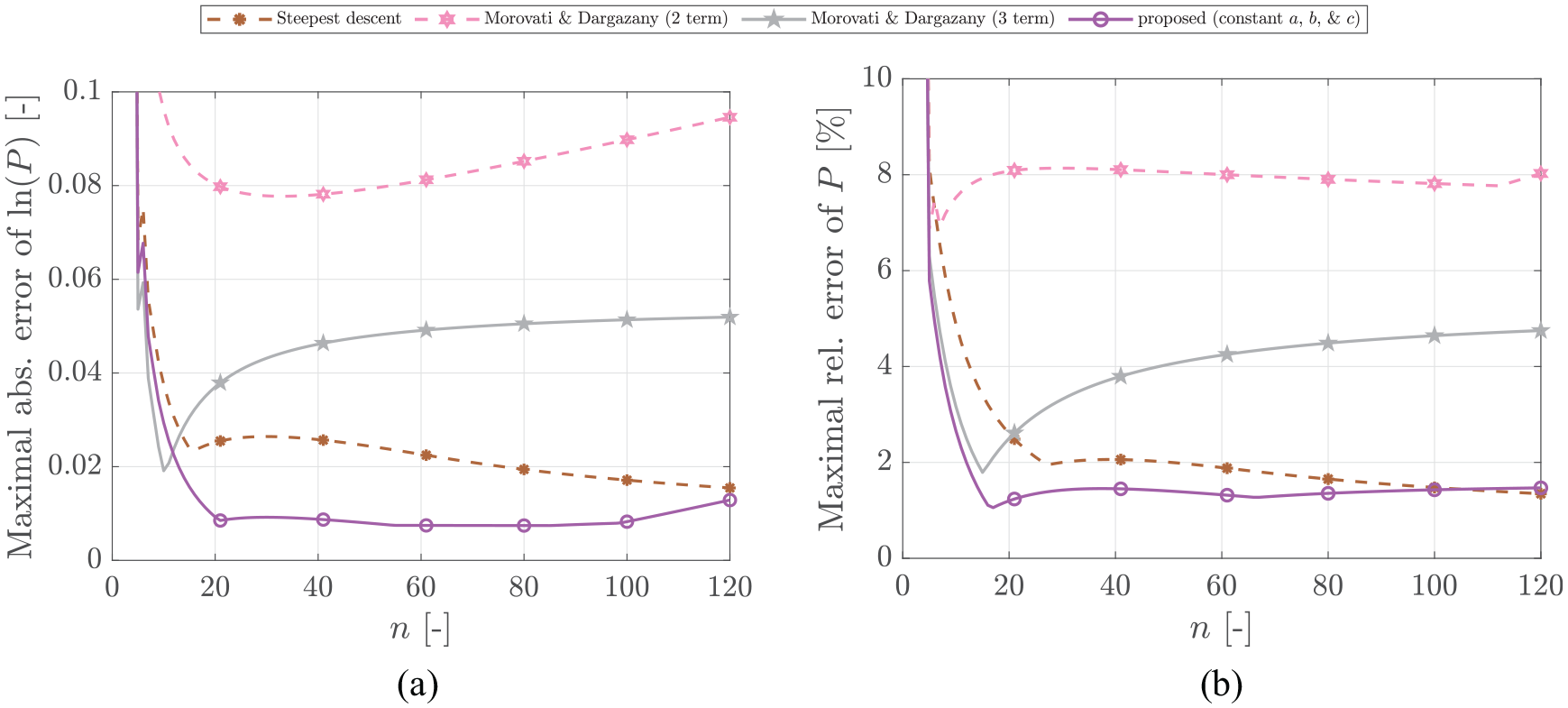
In a wide range of , the proposed Padé approximation-based approach is capable to provide stronger predictions in comparison to the others even when used with constant , , and . Figure 6 shows the resulting error, when utilized with averaged constant values for , , and . While the dependency on enhances the approximation for and , the effect is minimal for chains of medium size.
Comparison of the proposed Padé-based approximation as given in equation (24) with averaged parameters (, , and ) to the best approximations from the literature.
7. Conclusion
In the current work, we showed that most of the previously proposed approximations of the non-Gaussian probability distribution function (4) of the polymer chain length perform well only for a particular number of chain segments . The effect of on the approximation quality was investigated. Although some approximations outperform others, in the majority of cases, there is no clear superior approximation in the whole domain and for every considered error measure. The proper choice depends on the particular chain under investigation (determined by ) and the desired application. As a result, there exists no versatile unique approximation to meet all the requirements.
In this paper, we derived a family of approximations of the non-Gaussian probability distribution function on the basis of the Padé method. The developed method is able to incorporate the polymer chain length and can be tailored to any specific application. For example, formulation (24)–(27) provides the minimal absolute error of . In a wide range of , the proposed approach appears to be the most accurate in comparison to other approximations known from the literature. A simplified approach, which neglects the dependency on the number of chain segments , was shown to still outperform most of the alternative approximations.
The proposed approximation function is very robust for the modeling of polymer blends made up of short chains and also can minimize errors in simulating multimodal polymer networks which contain a wide range of polymer chain sizes [39]. Moreover, the non-Gaussian theory of polymer networks has been widely utilized to model the swelling behavior of cross-linked gels with finite extensibility of networks of high charge densities. The accuracy of such models can be considerably enhanced by the use of the proposed approach [40,41].
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iDs
Ehsan Darabi
Markus Hillgärtner
References
1.
EdwardsSF. Statistical mechanics with topological constraints: I. Proc Phys Soc1967; 91(3): 513.
2.
KuhnW. Beziehungen zwischen molekülgröße, statistischer molekülgestalt und elastischen eigenschaften hochpolymerer stoffe. Kollo Zeitsch1936; 76(3): 258–271.
3.
GuthE. Theory of filler reinforcement. J Appl Phys1945; 16(1): 20–25.
4.
KuhnWGrünF. Beziehungen zwischen elastischen konstanten und dehnungsdoppelbrechung hochelastischer stoffe. Koll Zeitsch1942; 101(3): 248–271.
5.
ArrudaEMBoyceMC. A three-dimensional constitutive model for the large stretch behavior of rubber elastic materials. J Mech Phys Solids1993; 41(2): 389–412.
6.
DargazanyRItskovM. A network evolution model for the anisotropic Mullins effect in carbon black filled rubbers. Int J Solids Struct2009; 46: 2967–2977.
7.
MieheCGöktepeSLuleiF. A micro–macro approach to rubber-like materials—part I: the non-affine micro-sphere model of rubber elasticity. J Mech Phys Solids2004; 52(11): 2617–2660.
8.
WuPVan Der, GiessenE. On improved network models for rubber elasticity and their applications to orientation hardening in glassy polymers. J Mech Phys Solids1993; 41(3): 427–456.
9.
WangMCGuthE. Statistical theory of networks of non-Gaussian flexible chains. J Chem Phys1952; 20(7): 1144–1157.
10.
FloryPJRehnerJJr.Statistical mechanics of cross-linked polymer networks I: rubber like elasticity. J Chem Phys1943; 11(11): 512–520.
11.
ItskovMEhretAEDargazanyR. A full-network rubber elasticity model based on analytical integration. Math Mech Solids2010; 15(6): 655–671.
12.
TreloarLRG. The physics of rubber elasticity. New York: Oxford University Press, 1975.
JerniganRFloryP. Distribution functions for chain molecules. J Chem Phys1969; 50(10): 4185–4200.
15.
RubinsteinMColbyRH. Polymer physics, vol. 23. New York: Oxford University Press, 2003.
16.
YamakawaH. Modern theory of polymer solutions. Manhattan, NY: Harper & Row, 1971.
17.
DillKBrombergS. Molecular driving forces: statistical thermodynamics in biology, chemistry, physics, and nanoscience. New York: Garland Science, 2012.
18.
RayleighL.XXXI. On the problem of random vibrations, and of random flights in one, two, or three dimensions. Lond, Edin Dubl Philos Mag J Sci1919; 37(220): 321–347.
19.
ChandrasekharS. Stochastic problems in physics and astronomy. Rev Mod Phys1943; 15(1): 1.
20.
TreloarLRG. The statistical length of long-chain molecules. Trans Faraday Soc1946; 42: 77–82.
21.
NagaiK. Elementary problems on the configurations of a linear high polymer. J Phys Soc Jpn1958; 13(8): 928–934.
22.
HsiungCHsiungHGordusAA. A closed general solution of the probability distribution function for three-dimensional random walk processes. J Chem Phys1961; 34(2): 535–546.
23.
De BruijnNG. Asymptotic methods in analysis, vol. 4. Chelmsford, MA: Courier Corporation, 1981.
24.
JamesHMGuthE. Theory of the elastic properties of rubber. J Chem Phys1943; 11(10): 455–481.
25.
ItskovMDargazanyRHörnesK. Taylor expansion of the inverse function with application to the Langevin function. Math Mech Solids2012; 17(7): 693–701.
26.
DarabiEItskovM. A simple and accurate approximation of the inverse Langevin function. Rheol Acta2015; 54(5): 455–459.
27.
JedynakR. New facts concerning the approximation of the inverse Langevin function. J Non-Newt Fluid Mech2017; 249: 8–25.
28.
KroegerM. Simple, admissible, and accurate approximants of the inverse Langevin and Brillouin functions, relevant for strong polymer deformations and flows. J Non-Newt Fluid Mech2015; 223: 77–87.
29.
MalacarneLMendesRLenziE, et al. A non-Gaussian model in polymeric network. Eur Phys J E2006; 20(4): 395–399.
30.
KhiêmVNItskovM. Analytical network-averaging of the tube model: rubber elasticity. J Mech Phys Solids2016; 95: 254–269.
31.
MorovatiVDargazanyR. Improved approximations of non-Gaussian probability, force, and energy of a single polymer chain. Phys Rev E2019; 99(5): 052502.
32.
FrobeniusG. Ueber Relationen zwischen den Näherungsbrüchen von Potenzreihen. J Die Reine Angew Math1881; 1881(90): 1–17.
33.
JacobiCGJ. Über die Darstellung einer Reihe gegebner Werthe durch eine gebrochne rationale Function. J Die Reine Angew Math1846; 1846(30): 127–156.
34.
PadéH. Sur la représentation approchée d’une fonction par des fractions rationnelles. Ann Sci l’École Norm Supér1892; 9: 3–93.
35.
WynnP. On the convergence and stability of the epsilon algorithm. SIAM J Num Anal1966; 3(1): 91–122.
36.
LevenbergK. A method for the solution of certain non-linear problems in least squares. Quart Appl Math1944; 2(2): 164–168.
37.
MarquardtDW. An algorithm for least-squares estimation of nonlinear parameters. J Soc Ind Appl Math1963; 11(2): 431–441.