
Abstract
Objective
To review the pharmacokinetic (PK)–pharmacodynamic (PD) profiles, disease setting, dosing, and safety of oral and parenteral hypomethylating agents (HMAs) for the treatment of myelodysplastic syndromes/neoplasms (MDS) and acute myeloid leukemia (AML), and to provide a multidisciplinary perspective on treatment selection and educational needs relating to HMA use.
Data Sources
Clinical and real-world data for parenteral decitabine and azacitidine and two oral HMAs: decitabine–cedazuridine (DEC-C) for MDS and azacitidine (CC-486) for AML maintenance therapy.
Data Summary
Differences in the PK–PD profiles of oral and parenteral HMA formulations have implications for their potential toxicities and planned use. Oral DEC-C (decitabine 35 mg and cedazuridine 100 mg) has demonstrated equivalent systemic area under the concentration-time curve (AUC) exposure to a 5-day regimen of intravenous (IV) decitabine 20 mg/m2 and showed no significant difference in PD. The AUC equivalence of oral DEC-C and IV decitabine means that these regimens can be treated interchangeably (but must not be substituted within a cycle). Oral azacitidine has a distinct PK–PD profile versus IV or subcutaneous azacitidine, and the formulations are not bioequivalent or interchangeable owing to differences in plasma time-course kinetics and exposures. Clinical trials are ongoing to evaluate oral HMA combinations and novel oral HMAs, such as NTX-301 and ASTX030.
Conclusions
Treatment with oral HMAs has the potential to improve quality of life, treatment adherence, and disease outcomes versus parenteral HMAs. Better education of multidisciplinary teams on the factors affecting HMA treatment selection may help to improve treatment outcomes in patients with MDS or AML.
Introduction
Myelodysplastic syndromes/neoplasms (MDS) and acute myeloid leukemia (AML) are two heterogeneous groups of hematopoietic cancers that share several common molecular defects. 1 Epigenetic dysregulation, leading to aberrant DNA hypermethylation, is a hallmark of both diseases. 2 Although mutations in epigenetic regulators, including those involved in DNA methylation, are largely similar between MDS and AML, the frequencies of these mutations differ. 3 Even in the absence of key driver mutations, hypermethylation of cytosine-phosphate-guanine–rich sites in promoter regions is implicated in both MDS and AML, resulting in the silencing of tumor suppressor genes and aberrant regulation of other genes involved in cellular proliferation, differentiation, and apoptosis.2,4 Considering the inherent reversibility of epigenetic modifications, the possibility of restoring normal DNA methylation has led to the pursuit of targeted epigenetic agents with hypomethylating potential. 4
Currently, two parenteral hypomethylating agents (HMAs) are approved for the treatment of MDS or AML and are the standard of care in these indications.5–8 Intravenous (IV) decitabine is approved for MDS in the United States (US) and for AML in the European Union (EU).5,6 Subcutaneous (SC) or IV azacitidine is approved in the US for MDS, and for both MDS and AML in the EU (Table 1).7,8
Characteristics of parenteral and oral HMAs.
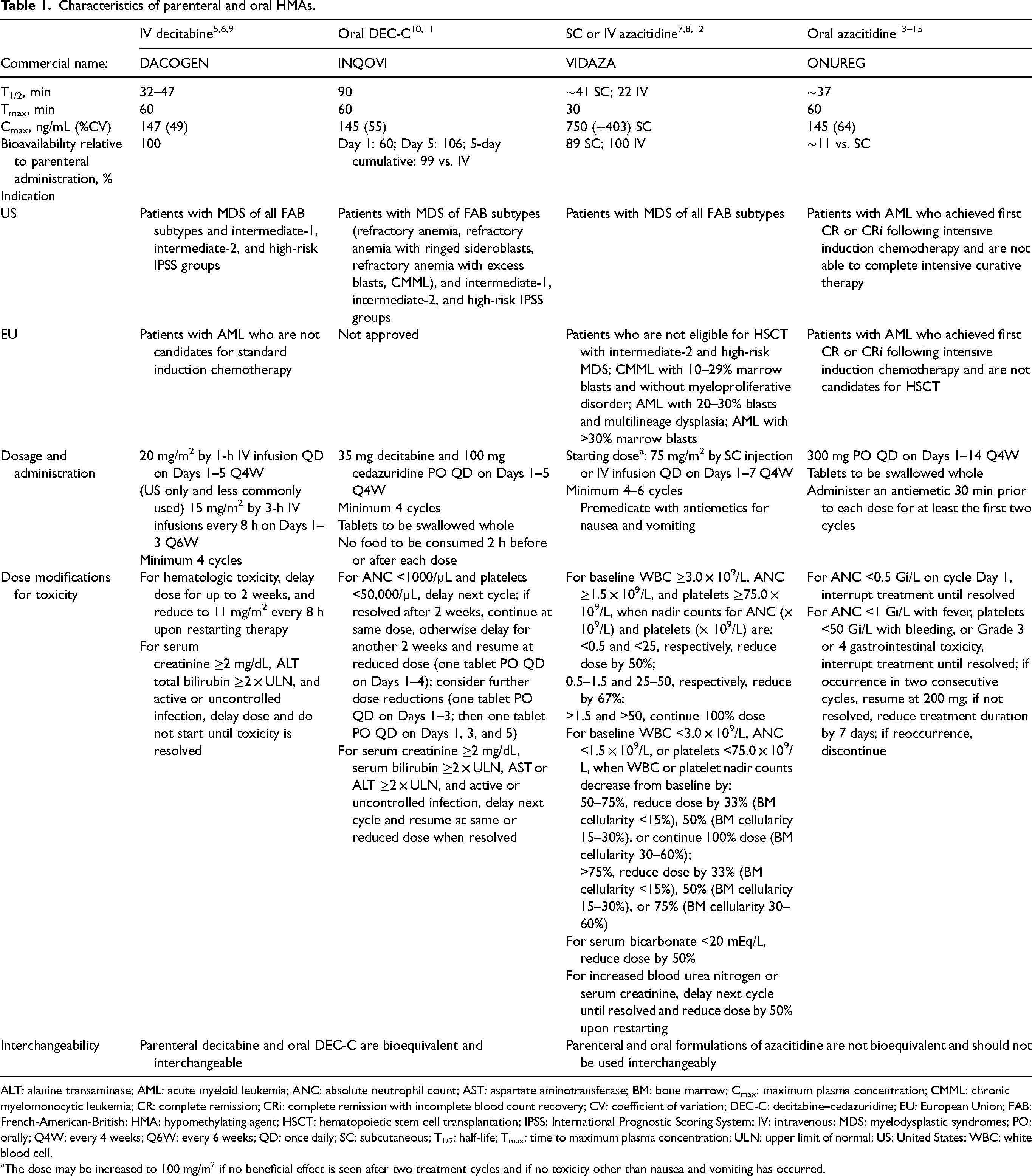
ALT: alanine transaminase; AML: acute myeloid leukemia; ANC: absolute neutrophil count; AST: aspartate aminotransferase; BM: bone marrow; Cmax: maximum plasma concentration; CMML: chronic myelomonocytic leukemia; CR: complete remission; CRi: complete remission with incomplete blood count recovery; CV: coefficient of variation; DEC-C: decitabine–cedazuridine; EU: European Union; FAB: French-American-British; HMA: hypomethylating agent; HSCT: hematopoietic stem cell transplantation; IPSS: International Prognostic Scoring System; IV: intravenous; MDS: myelodysplastic syndromes; PO: orally; Q4W: every 4 weeks; Q6W: every 6 weeks; QD: once daily; SC: subcutaneous; T1/2: half-life; Tmax: time to maximum plasma concentration; ULN: upper limit of normal; US: United States; WBC: white blood cell.
The dose may be increased to 100 mg/m2 if no beneficial effect is seen after two treatment cycles and if no toxicity other than nausea and vomiting has occurred.
Decitabine (5-aza-2’-deoxycytidine) and azacitidine (5-azacytidine) differ slightly in their chemical structure and mechanism of action: decitabine is incorporated exclusively into DNA, whereas most parenterally administered azacitidine is incorporated into RNA. 16 However, both are cytidine analogs that bind to and inhibit the activity of DNA methyltransferase 1 (DNMT1), which catalyzes the process of DNA methylation (Figure 1). Inhibition of DNMT1 results in global DNA hypomethylation and the reactivation of previously silenced genes, including critical tumor suppressor genes, which can be detected by the HMA-induced demethylation of long interspersed nuclear element 1 (LINE-1). Various additional antineoplastic mechanisms have been proposed for HMAs, including the reactivation of genes that mediate immune responses, cell cycle control, apoptosis, and DNA damage repair. 16
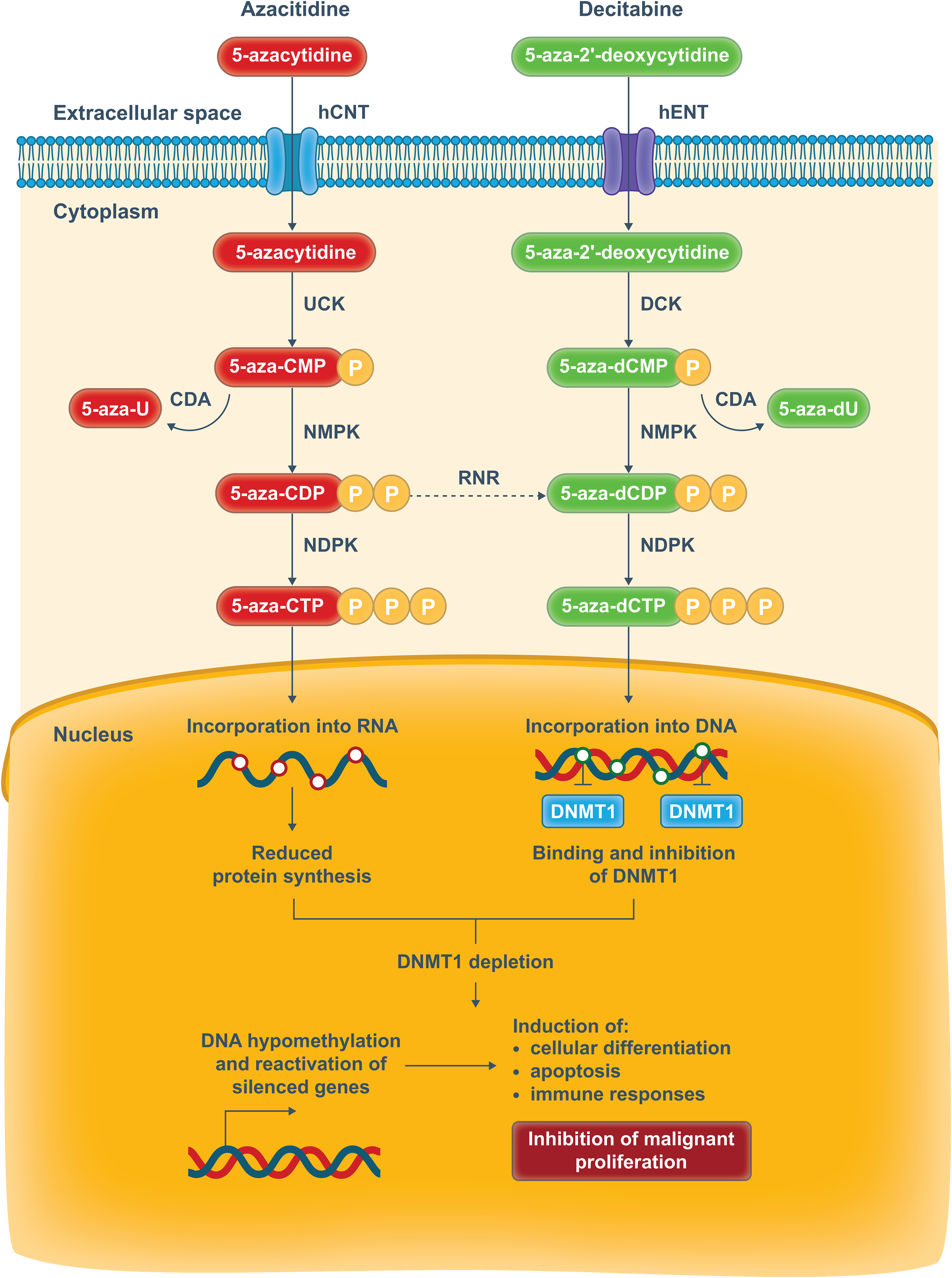
Cellular mechanism of action of the hypomethylating agents azacitidine and decitabine. 5-aza-U, 5-azacytidine-uridine; 5-aza-dU, 5-azacytidine-2'-deoxyuridine; CDP, cytidine diphosphate; CMP, cytidine monophosphate; DCK, deoxycytidine kinase; DNMT1, DNA methyltransferase 1; hCNT, human concentrative nucleoside transporter; hENT, human equilibrative nucleoside transporter; NDPK, nucleoside diphosphate kinase; NMPK, nucleoside monophosphate kinase; P, phosphate; RNR, ribonucleotide reductase; UCK, uridine-cytidine kinase.
In clinical trials, parenteral HMAs have been shown to improve response rates and outcomes such as health-related quality of life (QoL) in patients with MDS and AML, as well as delaying progression to AML in patients with MDS. 17 However, real-world outcomes among patients receiving parenteral HMAs appear less favorable than outcomes in clinical trials. 18 In a pivotal phase III trial in patients with higher-risk (HR-)MDS, azacitidine significantly prolonged overall survival (OS) versus conventional care (median: 24.5 vs. 15.0 months, respectively; p = 0.0001) at a median follow-up of 21.1 months. 19 However, this was the only clinical trial to report a median OS of 24.5 months. Data from comparative studies of azacitidine alone versus other therapies have reported a median OS of 11.8–16.9 months for azacitidine,20–23 and studies comparing azacitidine in combination with another agent versus azacitidine alone in patients with HR-MDS have reported a median OS of 13.4–19.1 months for azacitidine alone.24–28 Data from several real-world studies of azacitidine have reported a median OS range of 12.4–17.1 months.29–33
Real-world data suggest underuse of parenteral HMAs18,34; in a study based on data from the Surveillance, Epidemiology and End Results–Medicare database, 44% of patients with HR-MDS did not receive HMA therapy. 34 Furthermore, several studies have reported low rates of persistence with HMA therapy relative to guideline recommendations in patients with HR-MDS and AML.18,34 In the Surveillance, Epidemiology and End Results–Medicare study, 44% of HMA users were nonpersistent with therapy and received less than four cycles or had a gap of ≥90 days between consecutive cycles. 34 Optimal treatment outcomes with parenteral HMA formulations require long-term therapy involving a regular and uninterrupted schedule, and patients generally require a minimum of four to six cycles of five to seven consecutive daily doses per month.5,7 Reasons for early discontinuation of HMA therapy can be clinically driven, such as disease progression or toxicity, or nonclinically driven. 18 Parenteral routes of administration may be associated with psychological distress and can result in medical complications linked with venous access and adverse events (AEs), such as injection-site reactions and pain related to SC administration. Additionally, nonpersistence with HMA therapy may result from the need for regular visits to the infusion clinic and associated logistical and administrational challenges, particularly in more vulnerable patients with MDS, who often report impaired physical activity and may experience difficulties with instrumental activities of daily living.18,35 Not only can early discontinuation have a negative impact on treatment outcomes, including OS, but it can also result in significantly higher economic costs.18,36 The healthcare costs associated with the management of patients with MDS following early discontinuation of HMA therapy are approximately $77,000 per patient during the first 6 months and $130,000 for the subsequent 12 months. 37 Furthermore, access to and persistence with parenteral HMAs are influenced by socioeconomic and clinical factors, including age, marital status, and comorbidities (Table 2).18,34
Predictors of HMA use. 34
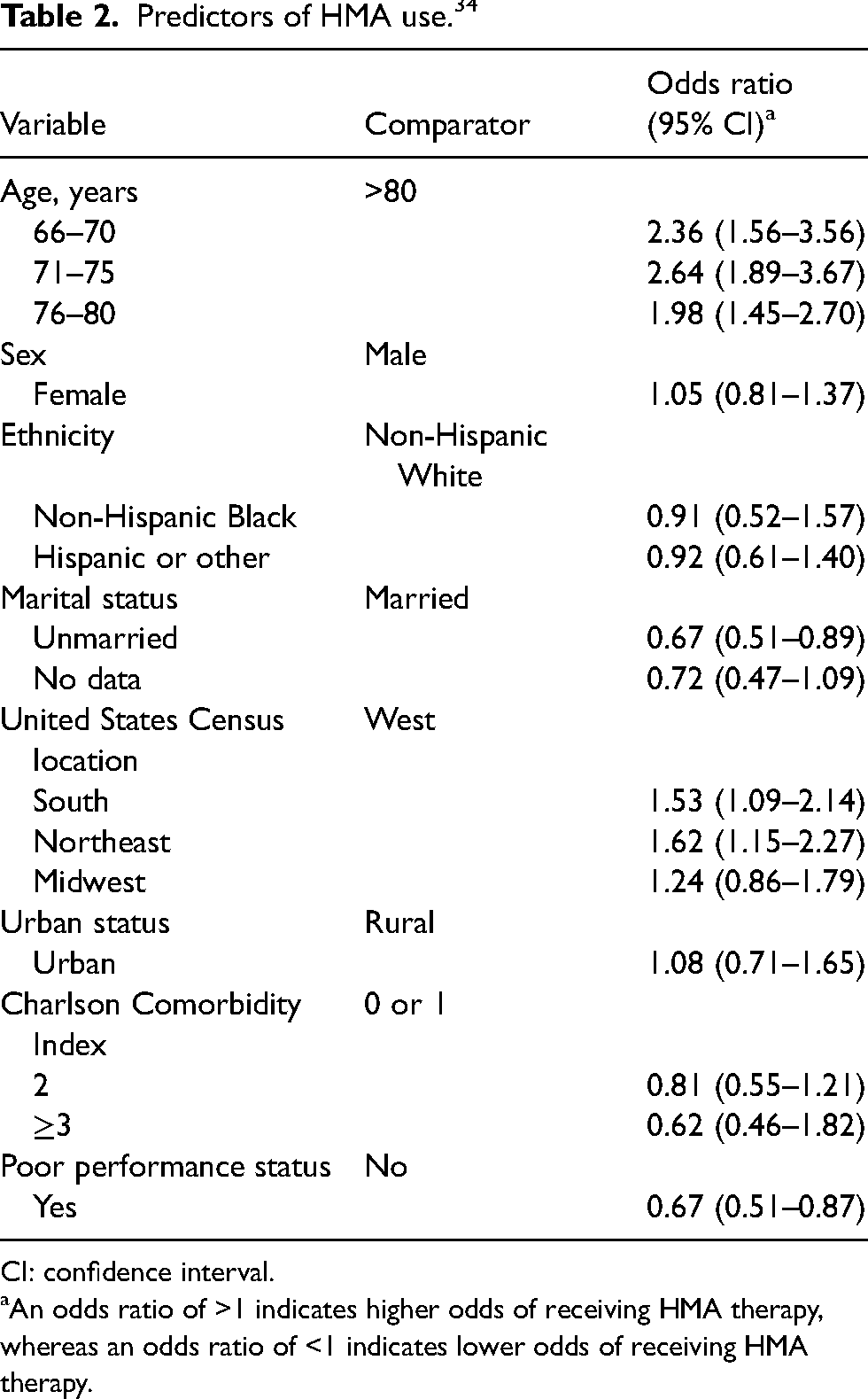
CI: confidence interval.
An odds ratio of >1 indicates higher odds of receiving HMA therapy, whereas an odds ratio of <1 indicates lower odds of receiving HMA therapy.
Oral HMA formulations have the potential to reduce the burden of therapy and improve adherence versus their parenteral counterparts. Two oral HMAs were approved in the US in 2020 for the treatment of MDS or AML: oral decitabine–cedazuridine (DEC-C) fixed-dose combination tablets for patients with previously treated and untreated, de novo and secondary MDS; and oral azacitidine (CC-486) tablets for patients with AML who achieve first complete remission (CR) or CR with incomplete blood count recovery (CRi) following intensive induction chemotherapy and are not able to complete intensive curative therapy (Table 1).10,13 These agents have shown efficacy in their respective indications and may improve persistence compared with parenteral HMAs by causing less disruption to daily life, reducing the stress and discomfort associated with SC and IV formulations, and presenting an alternative, oral option for patients who are unable to receive IV or SC infusions. 38 Thus, oral HMAs have the potential to reduce the treatment burden and decrease rates of early discontinuation, resulting in improved QoL, efficacy, and survival for patients with MDS and AML. 39
Differences in the pharmacokinetics (PK) and pharmacodynamics (PD) of oral versus parenteral HMA formulations (Table 1) have implications for their potential toxicities, mechanisms of action, and planned use. Moreover, oral DEC-C and oral azacitidine (CC-486) have distinct PK, PD, and safety profiles, resulting in differences in their approved indications. Understanding these differences is important for predicting potential toxicities and optimizing combination strategies.
The aim of this narrative review is to provide a multidisciplinary perspective on how the use of oral HMAs may reduce the burden associated with IV or SC formulations and potentially improve treatment outcomes. We will consider differences in the PK, PD, disease setting, dosing, and safety of the two available oral HMAs and parenteral HMA formulations and discuss recent advances in the development of HMAs and the role of HMAs according to risk. Practical considerations on the use of HMAs from a multidisciplinary team (MDT) perspective and the need for educational tools to increase understanding and the differences between oral and parenteral HMAs will also be discussed.
Parenteral decitabine versus oral DEC-C
Intravenous decitabine is approved in the US for the treatment of patients with MDS of all French-American-British (FAB) subtypes (refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia [CMML]) and intermediate-1, intermediate-2, or high-risk International Prognostic Scoring System (IPSS) groups; it is approved in the EU for patients with AML who are not candidates for standard induction chemotherapy (Table 1).5,6 Both the US Food and Drug Administration (FDA) and European Medicines Agency recommend a 5-day regimen of decitabine 20 mg/m2, administered as a once-daily (QD), 1-h IV infusion for 5 consecutive days in every 4-week treatment cycle.5,6 However, an earlier FDA-approved 3-day regimen, recommended in the US only, uses an IV infusion of decitabine 15 mg/m2 over 3 h repeated every 8 h for 3 days, and the cycle is repeated every 6 weeks. 5 A minimum of four cycles is recommended to be administered before lack of efficacy is established with either regimen.5,6
In patients with MDS or AML, the maximum plasma concentration (Cmax) of decitabine is generally observed at the end of IV infusion, when the steady-state concentration is reached.9,40 Decitabine is widely distributed over tissues and has a mean volume of distribution of 63–89 L/m2 at steady state.9,40 Elimination of decitabine is mainly metabolic, followed by renal clearance of degradation products. Results from a mass balance study using 14C-decitabine showed that 90% of the administered dose of decitabine (4% unchanged drug) was excreted in urine. 6 Similarly, a mass balance study of C-guadecitabine, a dinucleotide of decitabine and endogenous deoxyguanosine, showed that decitabine was mostly metabolized prior to excretion, with negligible (0.3%) unchanged drug detected in urine. 41 Deamination of decitabine is mediated by cytidine deaminase (CDA), which is highly expressed in the gut and liver.5,40 Decitabine is not a substrate of cytochrome P450 enzymes, has an elimination half-life of approximately 35–45 min in its IV form, and shows no systemic accumulation after repeat doses.5,9,40
Intravenous decitabine is generally well tolerated and has a manageable toxicity profile; the most common AEs in patients with MDS or AML are myelosuppressive toxicities (mainly Grade 3 or 4), particularly neutropenia and thrombocytopenia, and Grade 1 or 2 nonhematologic AEs, such as pyrexia, fatigue, and gastrointestinal toxicities (e.g., diarrhea, nausea).5,6,17,42
Early clinical experience with orally administered decitabine indicated that, when given alone, oral doses resulted in markedly lower plasma concentrations than with IV administration, and there was substantial variability in bioavailability (range: 4–14% relative to parenteral administration) but similarity in the short half-life (0.36–0.93 h). 43 These characteristics can be largely attributed to the rapid degradation of decitabine by CDA, which itself has high inter- and intraindividual variability in expression.39,44 To counteract this, oral decitabine was combined with the CDA inhibitor cedazuridine.
In preclinical studies, a sequential combination of cedazuridine and decitabine given 30 min apart achieved similar exposures to those of IV decitabine and induced demethylation of LINE-1, a surrogate marker of epigenetic modulation.45,46 In a subsequent phase I study in patients with MDS and CMML, concurrent administration of fixed-dose oral DEC-C significantly increased plasma bioavailability of decitabine, resulting in oral decitabine exposures equivalent to those with IV decitabine 20 mg/m2 administered for 5 consecutive days every 4 weeks. Oral decitabine 30 mg and 40 mg combined with cedazuridine 100 mg achieved area under the concentration-time curve (AUC) 5-day exposures of decitabine oral to IV ratios of 81% and 128%, respectively. Based on these findings, a combination of oral decitabine 35 mg plus cedazuridine 100 mg was selected as the recommended phase II dose (RP2D). 47
In a randomized phase II crossover study in patients with MDS and CMML, treatment with the RP2D of oral DEC-C resulted in similar systemic decitabine exposure and DNA demethylation, safety, and efficacy to IV decitabine. 48 Patients were randomized 1:1 to receive oral DEC-C QD on Days 1 through 5 in Cycle 1 and IV decitabine 20 mg/m2 on Days 1 through 5 in Cycle 2, or the reverse sequence, followed by oral DEC-C QD on Days 1 through 5 of each 28-day cycle in Cycle 3 and beyond. The mean ratios of oral to IV decitabine 5-day AUCs were 93.5% using separate capsules (n = 50) and 97.6% using a fixed-dose combination tablet (n = 30). Mean percentage LINE-1 demethylation differed by ≤1% between the oral and IV formulations. 48
The AUC equivalence (referred to hereafter as bioequivalence) of the oral DEC-C fixed-dose tablet and IV decitabine was subsequently confirmed in the phase III crossover study (ASCERTAIN) in patients with MDS and CMML.11,49 Overall, 133 patients were enrolled and randomized 1:1 to the same treatment schedule as described for the phase II crossover study. The primary endpoint of ratio of oral to IV decitabine 5-day AUC was 99%, indicating equivalent exposure (Table 1). Moreover, the difference in LINE-1 demethylation was <1%.11,49
Efficacy results from the phase II study reported an objective response rate (ORR) with oral DEC-C of 60% and CR, marrow CR (mCR), and hematologic improvement [HI] rates of 21%, 22%, and 16%, respectively; red blood cell (RBC) and platelet transfusion independence (TI) rates were both 50%, and median OS was 18.3 months. 48 In the phase III ASCERTAIN study, after a follow-up of 2.6 years, median OS for the total population was 31.8 months and leukemia-free survival was 31.7 months. 49 Of the 117 evaluable patients, 70% had a clinical response and 25% reached CR. The median durations of best response and CR were ∼1 year and ∼14 months, respectively. Among patients who were RBC or platelet transfusion dependent at baseline, 28/54 (52%) achieved RBC TI and 6/12 (50%) achieved platelet TI; 33% in each transfusion category achieved TI for ≥16 consecutive weeks. Twenty-seven patients (20%) proceeded to undergo hematopoietic stem cell transplantation (HSCT). 49
Overall, these efficacy data are consistent with previous clinical data for standard 5-day IV decitabine 20 mg/m2 dosing. In a multicenter study of IV decitabine in patients with MDS, the ORR (CR + mCR + partial response [PR] + HI) and CR rates were 51% and 17%, respectively, and the TI rates were 33–40%. 42 Safety findings with oral DEC-C were also like those observed with IV decitabine. The most common Grade ≥3 AEs in the phase II and III trials were hematologic toxicities (neutropenia: 38–52%, thrombocytopenia: 38–50%, anemia: 22–40%, leukopenia: 18–24%, febrile neutropenia: 14–29%). Furthermore, oral DEC-C was associated with similar incidence rates of gastrointestinal toxicities (which were predominantly Grade 1 or 2) versus IV decitabine.48–50
Based on demonstration of pharmacologic bioequivalence and comparable clinical efficacy to IV decitabine, oral DEC-C was approved by the FDA in July 2020 for the treatment of patients with MDS with the FAB subtypes of refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, and CMML, and IPSS intermediate-1, intermediate-2, and high-risk groups. 10 In a subanalysis based on TP53 mutational status of patients enrolled in the phase III ASCERTAIN study, median OS in patients with TP53-mutated MDS was 25.5 months compared with 33.7 months for those with TP53 wild-type gene status. Median OS in patients with monoallelic versus biallelic TP53-mutated MDS was 29.2 months versus 13.0 months. 51 In a separate subanalysis of 69 patients with IPSS low-risk or intermediate-1 MDS enrolled in the ASCERTAIN study, the ORR was 57%, the CR rate was 23%, and neither median leukemia-free survival nor OS had been reached after a median of 32 months of follow-up. 52 Low-dose oral DEC-C regimens for the treatment of lower-risk MDS are being evaluated in an ongoing phase I/II study (NCT03502668).
The National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology for MDS recommend oral DEC-C as a category 2A therapy for the management of MDS. 53 Intravenous decitabine can be substituted with oral DEC-C in patients with IPSS intermediate-1 and above, and oral DEC-C is recommended as a treatment option for the management of HR disease (IPSS-R intermediate-, high-, very high-risk disease) followed by allogeneic HSCT in transplant-eligible patients, for patients who are transplant ineligible, and for patients who relapse after allogeneic HSCT or have no response. 53 Oral DEC-C is also listed as an option for the management of lower-risk disease (IPSS-R very low-, low-, intermediate-risk disease) among patients with clinically relevant thrombocytopenia or neutropenia, for patients with symptomatic anemia with no del(5q) ± other cytogenetic abnormalities with ring sideroblasts <15% (or ring sideroblasts <5% with an SF3B1 mutation) and with serum erythropoietin >500 mU/mL pathway (poor probability to respond to immunosuppressive therapy), and certain patients with no response or intolerance to specific treatments. 53
In the approved indications, oral DEC-C should not be substituted for IV decitabine within a cycle. The recommended dosing of oral DEC-C is one tablet (35 mg decitabine and 100 mg cedazuridine) QD on Days 1 through 5 of each 28-day cycle for a minimum of four cycles. The European Medicines Agency has not yet formally approved oral DEC-C, but in July 2023 their Committee for Medicinal Products for Human Use adopted a positive opinion, recommending the granting of a marketing authorization for oral DEC-C for the initial treatment of adults with AML who are not candidates for standard induction chemotherapy. 54 In an interim analysis of the subpopulation of patients with AML in the phase III ASCERTAIN study (n = 87), oral DEC-C demonstrated bioequivalence to IV decitabine, with a decitabine oral over IV 5-day AUC of 99.6%. 55 Median OS with oral DEC-C was 7.9 months, CR rate was 21.8%, composite response rate (CR + CRi + PR) was 32.2%, and there were no noteworthy new safety signals. 55 Survival outcomes with oral DEC-C were comparable with those from phase III studies and real-world analyses of IV or SC HMAs for AML, such as the phase III ASTRAL-1 study in AML (N = 815), in which the median OS rates with IV or SC azacitidine or IV decitabine were 8.7 and 8.2 months, respectively. 56
Parenteral azacitidine versus oral azacitidine
Parenteral azacitidine is approved in the US for the treatment of patients with MDS of the following FAB subtypes: refractory anemia or refractory anemia with ringed sideroblasts (if accompanied by neutropenia or thrombocytopenia or requiring transfusions), refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and CMML (Table 1). 7 In the EU, parenteral azacitidine is indicated for patients with AML or IPSS-defined intermediate-2 and high-risk MDS who are not candidates for HSCT. 8 The recommended starting dose is 75 mg/m2 QD, administered by SC injection or IV infusion on Days 1 through 7 of each 28-day cycle. It is recommended that patients are treated for a minimum of four to six cycles (minimum six in the EU).7,8
Azacitidine is rapidly absorbed following SC administration in patients with MDS, with a Cmax of 750 ± 403 ng/mL reached after 0.5 h (Table 1).7,8,40 After SC administration, the absolute bioavailability of azacitidine relative to IV azacitidine is approximately 89%. As with decitabine, azacitidine is widely distributed in tissues; after IV infusion, the mean volume of distribution is reported to be 76 ± 26 L.7,8,12 Multiple dosing at the recommended dose does not result in drug accumulation with IV or SC administration. Like decitabine, azacitidine undergoes spontaneous hydrolysis and deamination by CDA, and its metabolism is not mediated by cytochrome P450 isoenzymes. The mean elimination half-life of azacitidine is approximately 41 min after SC administration and 22 min after IV infusion.7,8,12,40 Urinary excretion is the primary elimination route of azacitidine and its metabolites. Following IV administration, 85% of total radioactivity was recovered in urine, with fecal excretion accounting for <1% of administered radioactivity over 3 days. Mean excretion of radioactivity in urine after SC administration of 14C-azacitidine was 50%. A longer total radioactivity half-life of approximately 3.4–6.2 h, calculated based on 14C radioactivity, suggests the presence of circulating metabolites.7,40
The safety profile of IV or SC azacitidine in patients with MDS or AML is like that of IV decitabine. The most common AEs include hematologic toxicities such as anemia, thrombocytopenia, and neutropenia, and gastrointestinal AEs such as nausea, vomiting, diarrhea, and constipation; injection-site reactions are also common with the IV formulation.7,8,19
The oral (CC-486) and parenteral formulations of azacitidine have distinct PK profiles (Table 1) and should therefore be considered different drugs; in contrast to oral DEC-C and IV decitabine, the oral and parenteral formulations of azacitidine are not bioequivalent or interchangeable and should not be substituted.13,39 In a pilot study in four patients with AML, MDS, and solid tumors, oral azacitidine (CC-486) displayed rapid absorption following a single dose, and Cmax was reached within 1 h after dosing. Oral azacitidine was well tolerated at the 80-mg dose level and mean bioavailability was 17% relative to SC administration. 57
In a phase I/II dose escalation–expansion study designed to assess the maximum tolerated dose, safety, and PK–PD of a 7-day oral azacitidine (CC-486) administered after an initial cycle of SC azacitidine (75 mg/m2) in 41 patients with MDS, AML, or CMML, the plasma concentration-time profiles for oral and SC azacitidine were similar; however, a lower Cmax was observed with oral administration. Following a single 300-mg dose of oral azacitidine, the mean Cmax was 211 ng/mL versus 650 ng/mL for SC dosing. Relative to SC azacitidine, mean bioavailability with oral azacitidine was 11.5% at 300 mg and 12.8% at the maximum tolerated dose of 480 mg. The PK–PD analysis showed lower drug exposure and lower levels of global DNA hypomethylation with oral versus SC azacitidine, although this may have partly reflected previous exposure to SC azacitidine. The relatively poor bioavailability of oral azacitidine and lower DNA hypomethylation could not be overcome by increasing the dose, owing to the emergence of dose-limiting toxicity (Grade 3 or 4 diarrhea) at the highest dose. 15
Owing to these PK differences between the oral (CC-486) and parenteral formulations of azacitidine, ensuing development of oral azacitidine focused on evaluation of extended dosing schedules of either 14 or 21 days of treatment per cycle to prolong drug exposure and sustain therapeutic effects.2,15 Orally administered azacitidine has a short plasma half-life of approximately 30–40 min2,13,57; consequently, prolonging the duration of drug exposure resulted in more pronounced demethylation responses. 58 Analyses of the PK–PD of extended dosing schedules showed that administration of oral azacitidine (CC-486) 300 mg QD for 14 or 21 days resulted in sustained reductions in global DNA methylation in patients with MDS, CMML, or AML, and significant correlations were observed between azacitidine exposure and DNA hypomethylation.59,60 Compared with a 7-day regimen of SC azacitidine 75 mg/m2, cumulative exposures (AUC) per cycle with 14-day and 21-day oral dosing in patients with MDS were 38% and 57%, respectively. 60 Treatment with oral azacitidine using the 14-day or 21-day schedule showed similar efficacy (ORR: 36% vs. 41%; RBC TI: 31% vs. 38%) in patients with lower-risk (IPSS intermediate-1) MDS. In a separate phase II study, treatment with oral azacitidine 300 mg QD for 21 days of each 28-day cycle resulted in an ORR (CR + PR + mCR) of 32% in patients with MDS or CMML (n = 22) and an ORR (CR + PR + CRi) of 22% in patients with AML (n = 9). 61
Given the convenience of oral dosing and potential for long-term therapy, oral formulations are particularly appropriate for use as maintenance therapy. 2 Oral azacitidine (CC-486) was assessed as AML maintenance therapy in the randomized phase III QUAZAR AML-001 study in patients with AML who were in first remission after intensive chemotherapy and who were not candidates for HSCT (N = 472). 62 Median OS and relapse-free survival rates were both significantly longer with oral azacitidine than with placebo (24.7 vs. 14.8 months and 10.2 vs. 4.8 months, respectively; both p < 0.001), and improvements were observed across patient subgroups based on baseline characteristics. Health-related QoL was maintained throughout treatment in both the oral azacitidine and placebo arms. Oral azacitidine was well tolerated and had a similar safety profile to that of parenteral azacitidine. The most common AEs in both treatment arms were low-Grade gastrointestinal events, including nausea, vomiting, and diarrhea. Hematologic AEs (any Grade; Grade 3 or 4) in the oral azacitidine arm included neutropenia (44%; 41%), thrombocytopenia (33%; 22%), and anemia (20%; 14%). 62
Oral azacitidine (CC-486) has also been investigated in a phase III trial in patients with lower-risk MDS and RBC transfusion-dependent anemia and thrombocytopenia (N = 216). 63 Compared with placebo, significantly more patients in the oral azacitidine arm achieved the primary endpoint of RBC TI (31% vs. 11%; p = 0.0002), and the platelet HI rate was higher (24.3% vs. 6.5%). However, the trial was suspended owing to a higher death rate in the oral azacitidine group during the first 56 days (CC-486, n = 16; placebo, n = 6), which was mostly due to infections in patients who were severely neutropenic at enrollment. 63 The most common AEs in this study were gastrointestinal events, which were mostly Grade 1 or 2 and occurred more frequently in the oral azacitidine arm; neutropenia (47%) and thrombocytopenia (29%) were the most common Grade 3 or 4 treatment-emergent AEs. 63
Based on the findings of the phase III QUAZAR AML-001 trial, oral azacitidine (CC-486) was approved in the US in September 2020 and in the EU in June 2021 as maintenance therapy for the treatment of adult patients with AML who achieved first CR or CRi following intensive chemotherapy and who are ineligible for HSCT (Table 1).13,14 The recommended dosage is 300 mg orally QD on Days 1 through 14 of each 28-day cycle. As highlighted in the US and EU labels, oral azacitidine should not be used interchangeably with IV or SC azacitidine due to differences in the indications, dosage, and exposure, and healthcare professionals are advised to verify the name of the medicinal product, dose, and administration route prior to use.13,14
Further studies are needed to determine whether treatment with oral azacitidine is appropriate for patients with HR-MDS, due to the increased risk of early mortality. The current US and EU indications are limited to maintenance therapy in patients with AML,13,14 and it is explicitly stated in the US prescribing information that the use of oral azacitidine to treat patients with MDS is not recommended outside of controlled trials. 13
Advances in the development of HMAs
In addition to oral DEC-C and azacitidine, novel oral HMAs are in development for the treatment of MDS and/or AML (Table 3). These include NTX-301, a novel oral DNMT1 inhibitor, and ASTX030, an oral combination of azacitidine and cedazuridine that, unlike CC-486, is intended to have a similar plasma AUC exposure profile as IV azacitidine. Another active area of investigation is the combination of oral HMAs with other agents that have shown, or have potential for, clinical activity in patients with MDS or AML. Planned or ongoing trials of fully oral combinations (Table 3) include a phase I trial of oral DEC-C in a doublet regimen with the Janus kinase 1–selective inhibitor itacitinib for the treatment of MDS and relapsed or refractory overlap syndromes, phase I/II trials investigating oral DEC-C in a doublet combination with the oral B-cell lymphoma 2 inhibitor venetoclax for the treatment of high-risk MDS, CMML or AML, phase I/II studies of oral DEC-C in doublet or triplet combinations with ivosidenib, enasidenib, or gilteritinib plus venetoclax, or venetoclax plus the menin inhibitor SNDX-5613, and phase I/II studies of oral azacitidine in combination with venetoclax for HR-MDS or AML. Fully oral combination therapies may provide the benefits discussed previously, such as the potential to reduce the treatment burden and enhanced adherence compared with parenteral therapies.
Novel oral HMAs and fully oral HMA combination regimens under clinical investigation for the treatment of MDS and/or AML.
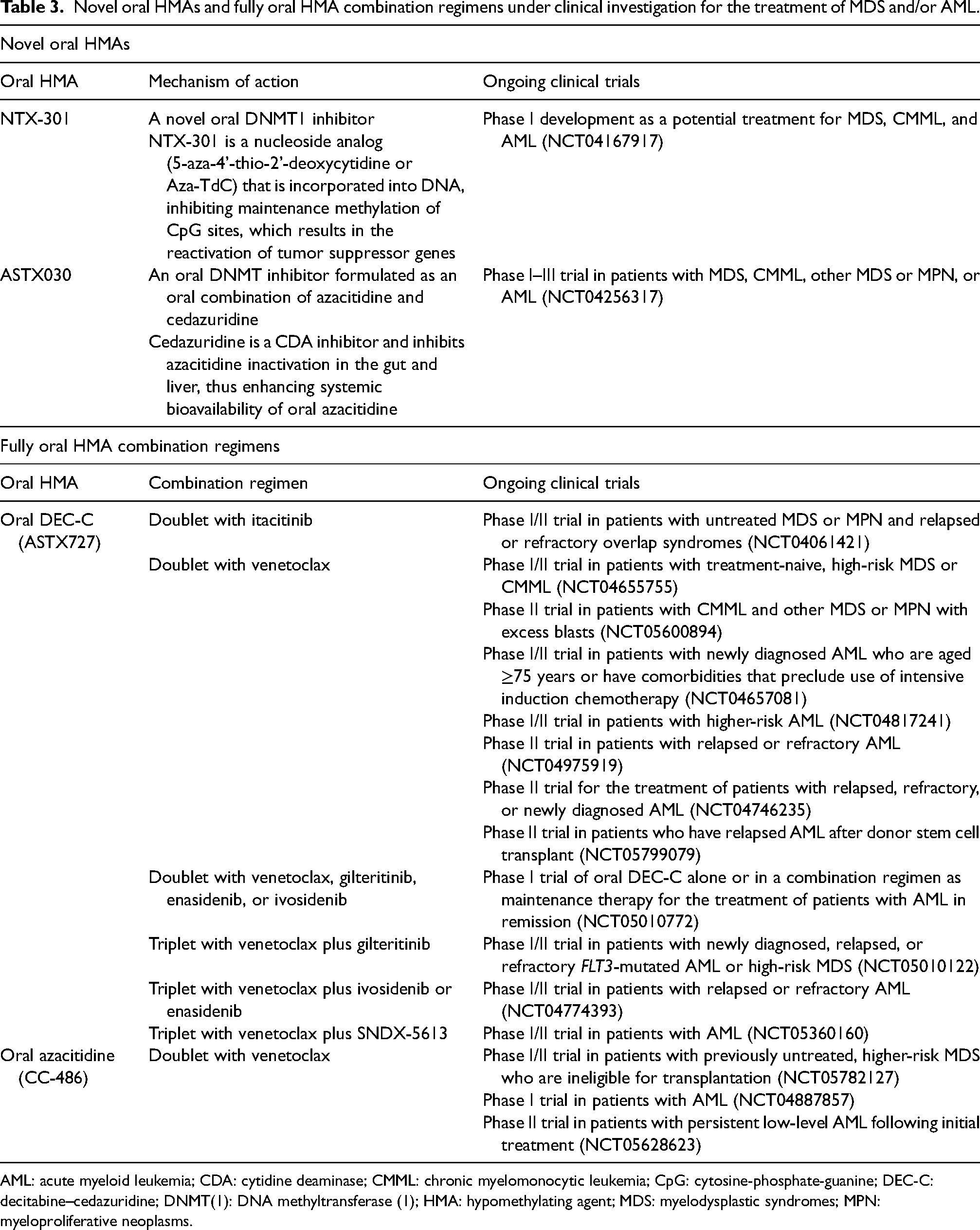
AML: acute myeloid leukemia; CDA: cytidine deaminase; CMML: chronic myelomonocytic leukemia; CpG: cytosine-phosphate-guanine; DEC-C: decitabine–cedazuridine; DNMT(1): DNA methyltransferase (1); HMA: hypomethylating agent; MDS: myelodysplastic syndromes; MPN: myeloproliferative neoplasms.
Other potential combination partners for oral HMAs include IV agents such as the anti–T-cell immunoglobulin and mucin domain 3 (TIM3) monoclonal antibody (mAb) sabatolimab (NCT04878432, NCT03066648, NCT03946670), the anti-CD47 mAb magrolimab (NCT04778410, NCT05835011), the anti–programmed death-ligand 1 mAb durvalumab (NCT02281084), and the anti–interleukin-8 mAb BMS-986253 (NCT05148234). Notably, in the phase III PANTHER trial, patients with HR-MDS, CMML, or AML failed to benefit from a combination of the NEDD8-activating enzyme inhibitor pevonedistat plus IV or SC azacitidine versus IV or SC azacitidine alone, potentially owing to a heterogeneous patient population (NCT03268954). 24 Subsequently, a phase II trial of IV pevonedistat plus oral DEC-C in patients with HR-MDS was terminated by its sponsor (NCT04985656).
In addition to combination approaches, oral formulations with predictable PK profiles are particularly well suited to use in alternative, metronomic dosing schedules, which may be appropriate for treating chronic and lower-risk conditions. Indeed, the use of regular much lower doses of IV decitabine (0.1–0.2 mg/kg/day administered 1–3 days per week) relative to the FDA-approved dose (15–20 mg/m2/day for 3–5 days every 28 days) have been shown to reduce cytotoxicity and increase exposure time for DNMT1 depletion. 64 Studies investigating alternative dosing regimens of oral HMAs are ongoing across a spectrum of myeloid malignancies. The phase I ASTX727-03 study (NCT03502668) is a multicenter, open-label study of various low-dose oral DEC-C regimens designed to assess safety, PD, PK, and hematologic response in patients with IPSS low-risk or intermediate-1 MDS. The RP2D (decitabine 10 mg plus cedazuridine 100 mg QD for 5 days) is currently being compared with decitabine 35 mg plus cedazuridine 100 mg for 3 days in a 28-day cycle in the ongoing phase II part of the study.
Multidisciplinary perspective: Considerations for treatment selection
Unlike parenteral HMAs, which require administration by skilled staff at a healthcare facility, oral DEC-C and azacitidine can be taken by patients at home and without concerns over injection-site reactions, thereby increasing patient autonomy and reducing healthcare costs associated with frequent clinic visits and management of injection- or infusion-related AEs. 2 However, like parenteral HMAs, sustained, on-schedule treatment with oral HMAs and ongoing supportive care is critical to ensuring optimal therapeutic efficacy 39 ; for example, it is important that patients are monitored as usual in the first two cycles prior to obtaining a response. It is therefore important to consider the patient's fitness and suitability for treatment, as well as factors that may affect treatment adherence or access to treatment, such as availability of transportation to the clinic. Other practicalities that need to be considered include monitoring AEs and making the associated dose modifications.
Survey data from patients receiving oral DEC-C suggest it has the potential to reduce treatment burden relative to IV or SC HMAs.38,65 Trends from real-world data among patients receiving oral DEC-C suggest comparable or improved compliance compared with IV or SC HMAs. An analysis of HMA claims between patients receiving oral DEC-C at home versus IV or SC treatment in the clinical setting suggested comparable or improved compliance with oral DEC-C and a possible advantage in reducing treatment burden. 66 In a survey to assess patient perspectives among 150 patients receiving oral DEC-C during 2021–2022, it was found that treatment had very little to no impact on activities of daily living for most patients, it improved QoL relative to IV or SC HMAs, and it had the potential to reduce visits to healthcare facilities compared with IV or SC HMAs. 65 Additionally, the advantage of reduced visits is magnified in patients who respond and are treatment-free for many months or years.
Practical considerations on the use of both parenteral and oral HMAs for hematology MDTs in routine practice are summarized in Table 4. Before initiating treatment, a designated MDT of healthcare professionals, including clinicians, clinical pharmacists, nurse practitioners (NPs), and physicians’ assistants, should consider the patient's suitably for HMA treatment on a case-by-case basis, considering a range of factors, such as comorbidities (e.g., preexisting cytopenias), current treatments (e.g., erythropoiesis-stimulating agents, blood transfusions), and patient preferences. 67
Considerations on the use of HMAs for hematology MDTs.

AE: adverse event; AML: acute myeloid leukemia; EU: European Union; HMA: hypomethylating agent; MDS: myelodysplastic syndrome; MDT: multidisciplinary team; NP: nurse practitioner; US: United States.
Clinicians, pharmacists, and NPs also closely monitor patients’ tolerance of therapy and monitor for AEs and signs or symptoms of any treatment complications that may necessitate dose reductions or delays. Additionally, ongoing supportive care is often required to manage HMA-associated AEs, such as antiemetics for nausea and vomiting, hematopoietic growth factors and antibiotics for infection prophylaxis or treatment, and blood product transfusion support for managing cytopenias.5–8,10,13,14
Nonpersistence with HMAs is associated with a poorer prognosis, more rapid disease progression (including progression to AML), and higher healthcare resource use.18,34,36 Therefore, it is important to understand the factors associated with treatment persistence, including the level of caregiver support and patients’ expectations of and willingness to accept toxicity with HMAs. Moreover, educating patients that MDS itself can be life-threatening, even without progression to AML, may facilitate better treatment persistence. 68
Lastly, access to HMA treatment, whether parenteral or oral, depends on the approved indication(s) and availability of insurance or reimbursement across different geographic regions. Out-of-pocket costs for patients and availability of patient assistance programs or grants may also vary depending on the specific therapeutic agent and on whether the administration is oral or IV, thus influencing treatment decisions. Access to parenteral HMAs (requiring injection or infusion) may be influenced by their availability and level of expertise at the treatment center, as well as the patient's distance from the clinic, their access to transportation, and socioeconomic factors that may influence their access to care.
Multidisciplinary perspective: Educational needs
Educational needs on the understanding and differentiation of oral HMAs are summarized in Table 5. It is important to ensure that the MDT can make evidence-based decisions regarding the use of HMAs based on PK–PD, disposition, and tolerability, and that they consider the patient perspective, dose adjustments or delays, and monitoring guidelines. Considering the two oral HMAs discussed in this review, the oncology clinical pharmacist is often the MDT member who is best suited to educate other healthcare providers on details of the differing registrational paths, study designs, PK profiles, and guidelines around interchangeability with parenteral formulations. In addition to patient selection, the MDT may be involved in providing education and training for other healthcare providers (e.g., registered nurses and social workers), particularly for community practices. This includes ensuring that all members of the MDT understand the importance of treatment persistence and factors that promote or interfere with adherence and have an appreciation of the patient's perspective on the goals and burden of treatment. Education of patients on proper use of treatment and the importance of prolonged and continuous treatment, and patient counseling on what to expect in terms of potential AEs, is also recommended. Educational materials that discuss this information should be provided when a prescription is first initiated to set treatment expectations with the patient and encourage timely self-reporting of symptoms to ensure relevant dose adjustments.
Educational needs.

AE: adverse event; HMA: hypomethylating agent; MDT: multidisciplinary team; PD: pharmacodynamics; PK: pharmacokinetics.
Conclusions
Hypomethylating agents are a mainstay of treatment for MDS and patients with AML who are not eligible for intensive chemotherapy. Compared with parenteral HMAs, the oral HMAs DEC-C and azacitidine have the potential to improve convenience, QoL, and treatment persistence for patients with MDS or AML, respectively. However, although both agents have received regulatory approval, they have different PK–PD profiles that render them distinct in their potential toxicities and clinical use. Oral DEC-C is bioequivalent to and can be used interchangeably with, IV decitabine in patients with MDS, as reflected in the approved indications. Conversely, oral (CC-486) and parenteral formulations of azacitidine have distinct PK profiles and are considered different drugs that should not be substituted for one another.
Currently, DEC-C is the only oral HMA that is approved for the treatment of MDS; oral azacitidine (CC-486) is indicated for patients with AML in remission after intensive induction chemotherapy who are not candidates for curative therapy, and its use for treatment of MDS is not recommended outside of controlled trials owing to the higher death rate observed in the oral azacitidine group in the phase III QUAZAR AML-001 study. Research into alternative dosing schedules and the combination of oral HMAs with other potential treatments for MDS or AML is ongoing, including evaluation of a combination of oral azacitidine and cedazuridine (ASTX030) based on the model for oral DEC-C, whereby coadministration with cedazuridine may potentially enhance the systemic bioavailability of oral azacitidine.
Several factors should be considered by clinicians, pharmacists, and NPs when making evidence-based decisions regarding HMA use, and pharmacists are often the best-positioned member of the MDT for providing education on these. Accordingly, better education of MDTs on the understanding and differentiation of oral HMAs, as well as the importance of treatment persistence and how best to manage toxicities, may help to improve treatment outcomes and QoL for eligible patients with MDS and AML. Additional clinical and real-world data are required to support the wider use of oral HMAs.
Footnotes
Acknowledgments
Medical writing assistance was provided by Farhana Burnett, of Envision Pharma Group, funded by Taiho Oncology, Inc. All authors have authorized the submission of this manuscript via third party and have approved the Declaration of Conflicting Interests, Funding, and Author Contributions statements.
Author contributions
All authors contributed to the concept and design of the work. All authors critically reviewed and edited the manuscript and approved the final version of the manuscript.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JK-S is Chair of the Summit & Julie Shah Foundation; is a member of Clinica Latina Board and Columbus Free Clinic Board; and has received honoraria or support for attending meetings from Hematology/Oncology Pharmacy Association and Vizient. AMZ is a Leukemia and Lymphoma Society Scholar in Clinical Research; has received research funding (institutional) from Abbvie, Aprea, Astex, BeyondSpring, Boehringer-Ingelheim, Celgene/BMS, Foran, Geron, Incyte, Kura, Medimmune/AstraZeneca, Novartis, Pfizer, Shattuck Labs, and Takeda; has received honoraria from AbbVie, Agios, ALX Oncology, Amgen, Astellas, BioCryst, Boehringer-Ingelheim, Celgene/BMS, Chiesi, Daiichi Sankyo, Epizyme, Genentech, Geron, Gilead, Incyte, Ionis, Janssen, Jazz, Kura, Mendus, Notable, Novartis, Orum, Otsuka, Pfizer, Regeneron, Schrodinger, Seattle Genetics, Servier, Syndax, Syros, Taiho Oncology, Takeda, Tyme, and Zentalis; and has served on clinical trial committees for Abbvie, ALX Oncology, BioCryst, Celgene/BMS, Geron, Gilead, Kura, Novartis, and Syros. RH and LB have no relevant conflicts of interest.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by Taiho Oncology, Inc.