
Abstract
Objective
Oral tyrosine kinase inhibitors (TKIs) are first line therapy for chronic myeloid leukemia (CML). A complete cytogenetic response (CCyR) correlates with increased overall survival, however only 66%–88% of patients achieve CCyR after one year of TKI treatment. Because TKI therapy alone cannot eliminate CML stem cells, strategies aimed at achieving faster and deeper responses are needed to improve long-term survival. Metformin is a widely prescribed glucose-lowering agent for patients with diabetes and in preclinical studies, has been shown to suppress cell viability, induce apoptosis, and downregulate the mTORC1 signaling pathway in imatinib resistant CML cell lines (K562R). This study aims to investigate the utility of metformin added to TKI therapy in patients with CML.
Data Sources
An observational study at an academic medical center (Salt Lake City, UT) was performed for adults with newly diagnosed, chronic-phase CML to evaluate attainment of CCyR from TKI therapy with or without concomitant metformin use. Descriptive analyses were used to describe baseline characteristics and attainment of response to TKI therapy.
Data Summary
Fifty-nine patients were evaluated. One hundred percent (5 of 5) in the metformin group and 73.6% (39 of 54) in the non-metformin group achieved CCyR. Approximately 20% of patients in both groups relapsed (defined by a loss of CCyR during study) after a median 34.5 months of follow-up.
Conclusions
Future research is warranted to validate these findings and determine the utility of metformin added to TKI therapy.
Keywords
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm accounting for approximately 15% of adult leukemias with an expected 8450 new diagnoses in the United States in 2020. 1 Central pathogenesis of CML is defined by the presence of the Philadelphia chromosome (Ph+), which results from a translocation between the Abelson murine leukemia (ABL1) gene and breakpoint cluster region (BCR) on chromosomes 9 and 22, respectively.2,3 The expression of BCR-ABL1 fusion protein results in deregulation of tyrosine kinase activity and constitutively active downstream intracellular signaling, leading to aberrant leukemogenesis. 4
Current treatment paradigm
For the majority of patients, tyrosine kinase inhibitor (TKI) therapy has changed CML into a manageable, chronic condition requiring frequent initial monitoring (every 3 months for the first 12 months) to assess response. Response milestones, in the order in which they are achieved, include normalization of peripheral blood counts (hematologic response; 1 log reduction in BCR-ABL1 copies), reduction in Ph+ chromosome metaphases (cytogenetic response; 2 log reduction in BCR-ABL1 copies), and decline in the number of BCR-ABL1 mRNA transcripts (molecular response; 3 log reduction in BCR-ABL1 copies). 2 Normalization of peripheral counts typically occurs within the first two to four weeks of TKI initiation when the bone marrow regains normal hematopoietic function. The next milestone, a complete cytogenetic response (CCyR; BCR-ABL1 < 1% per International Scale 5 ) to be achieved within 12 months of starting first-line therapy, has been correlated with improved overall survival.2,6,7 Once CCyR is achieved, ongoing molecular response assessment is required to detect BCR-ABL1 transcripts despite elimination of Ph+ chromosome metaphases. A deeper, molecular response is then assessed every 3 months with the goal of achieving specific milestones to guide both dose reductions and treatment-free remission (TFR) trials. 2
Standard of care for chronic-phase CML prior to the early 2000s produced dismal CCyR rates of only 10%. 8 Since the introduction of BCR-ABL1-directed TKIs, CCyR rates have increased to 66%–88% after just one year of therapy.9–13 Despite these advances, clearly not all patients attain CCyR. Numerous variables account for suboptimal response rates, and while the most important remains adherence to TKI therapy, 14 perfect adherence does not guarantee response. Therefore, the ability to increase the percentage of patients who attain a CCyR within 12 months of TKI therapy initiation is crucial for improved overall outcomes.
Opportunity for improvement
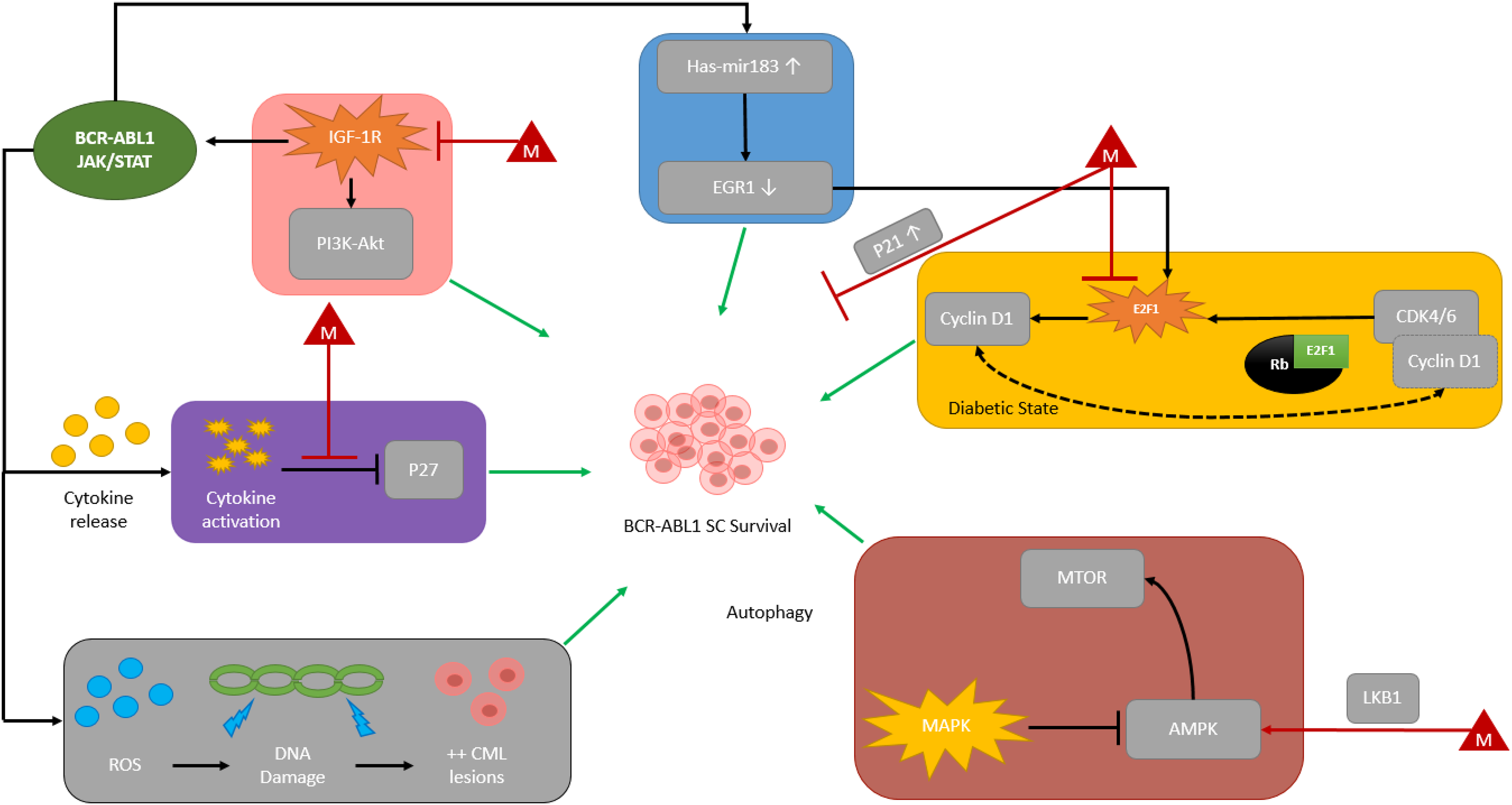
Although TKIs have forever changed CML management, the development of resistance, manifesting most commonly as amino acid substitutions (single, or compound) in the ATP-binding pocket, is still a relatively common occurrence.15,16 Because TKIs alone have not been shown to fully eradicate CML stem cells they cannot be considered curative.17–21 Additionally, TFR trials are not always successful; TFR failures have been observed as far out as 6 years post-TKI discontinuation, a timepoint where certain other hematologic malignancies such as acute myeloid leukemia and many lymphomas may be considered cured. Thus, novel strategies aimed at interfering with molecular-level resistance mechanisms employed by CML stem cells are still needed. In recent years, a growing body of in vitro and murine data has investigated the potential antileukemic activity of antidiabetic agents. Metformin is a widely prescribed glucose-lowering agent for patients with diabetes and in preclinical studies, has been shown to suppress cell viability, induce apoptosis, and downregulate the mTORC1 signaling pathway in imatinib resistant CML cell lines (K562R). 4 Previously proposed molecular pathways for BCR-ABL1 stem cell survival and possible metformin-induced impact are shown in Figure 1.4,22–42 Synergism between TKIs and metformin, should it exist, could have dramatic clinical implications on CML research. Therefore, we conducted a single-center, observational study to evaluate CCyR in patients with CML on TKI therapy also taking metformin.
Methods
Study design
This observational study was conducted at Huntsman Cancer Institute in Salt Lake City, Utah. Patients ≥ 18 years old diagnosed with CML who initiated TKI therapy on or after May 2014 (date of electronic medical record implementation) to January 2019 were queried for data analysis using the local institution's cancer clinical research database. The study included patients with newly diagnosed, chronic-phase CML who were treated with TKI therapy. Groups were divided based on concomitant use of metformin (metformin group) or not (non-metformin group). Patients were excluded if assessment of CCyR was unattainable (e.g., patient was deceased prior to assessment of CCyR or had not been on treatment for at least one year from TKI initiation). This study was approved by the local Institutional Review Board. Research reported in this publication utilized the Research Informatics Shared Resource at Huntsman Cancer Institute at the University of Utah and was supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA042014. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
The primary endpoint sought to determine the proportion of patients who achieved CCyR on TKI therapy with or without metformin at one year. At our institution, BCR-ABL1 fusion transcripts were quantified according to standard of care and reported using the international scale (IS). CCyR was defined as the absence of Ph+ chromosome-positive metaphases from the bone marrow or a BCR-ABL1 transcript level of ≤ 1%. Secondary endpoints included proportion of patients who achieved major molecular response (MMR) or complete molecular response (CMR), time to molecular response, rate of relapse, characterization of BCR-ABL1 mutation profiles (Y253H, E255K/V, F359V/C/I, F317L/V/I/C, T315A, V299L, or T315I), rates of stem cell transplant, and initial prescribed daily dose of metformin. MMR and CMR were defined as BCR-ABL1 transcript levels of ≤ 0.1% and ≤ 0.0032%, respectively. Relapse was defined as loss of complete cytogenetic response.
Data collection
Data were collected from the medical record and the institution's internal cancer clinical research database utilizing a standardized case report form in REDCap®. Data included baseline demographics (age, gender, and race) and CML history (date of CML diagnosis if known, Sokal risk score, mutation status, and name of first-line TKI therapy). For patients with a history of diabetes, data collected included date of diabetes diagnosis (if known for those patients with diabetes), hemoglobin A1c, and class of glucose-lowering agents used. Initial dose of metformin was recorded for patients who were on metformin. Response milestones and characterization of relapse were also captured. Descriptive analyzes were used to describe all endpoints. Time to molecular response rates were calculated using Kaplan-Meier method. Analyses were performed by using JMP Pro® version 14 (SAS Institute, Cary, NC).
Results
Baseline characteristics
One-hundred and ten patients were identified for inclusion based on the institution's internal clinical cancer research database (Figure 2). Fifty-one patients were subsequently excluded due to initiating a TKI prior to May 2014 (n = 41), missing data (n = 4), or an initial diagnosis of non-chronic phase CML (n = 6). A total of 59 patients were included in this study, 5 on TKI therapy and metformin and 54 on TKI therapy without metformin. Baseline characteristics were well balanced between both groups (Table 1). Median age at diagnosis was 54, over half of the total population (57%) was male, and approximately 90% were Caucasian. There was a larger distribution in CML risk stratification in the non-metformin group, with 38.9% (n = 21) having low risk disease compared to 60% (n = 3) in the metformin group. In both groups, the most common TKI therapy initiated was imatinib (55.9%) followed by dasatinib (23.7%). All patients in the metformin group had a diagnosis of diabetes compared to 14.8% in the non-metformin group.
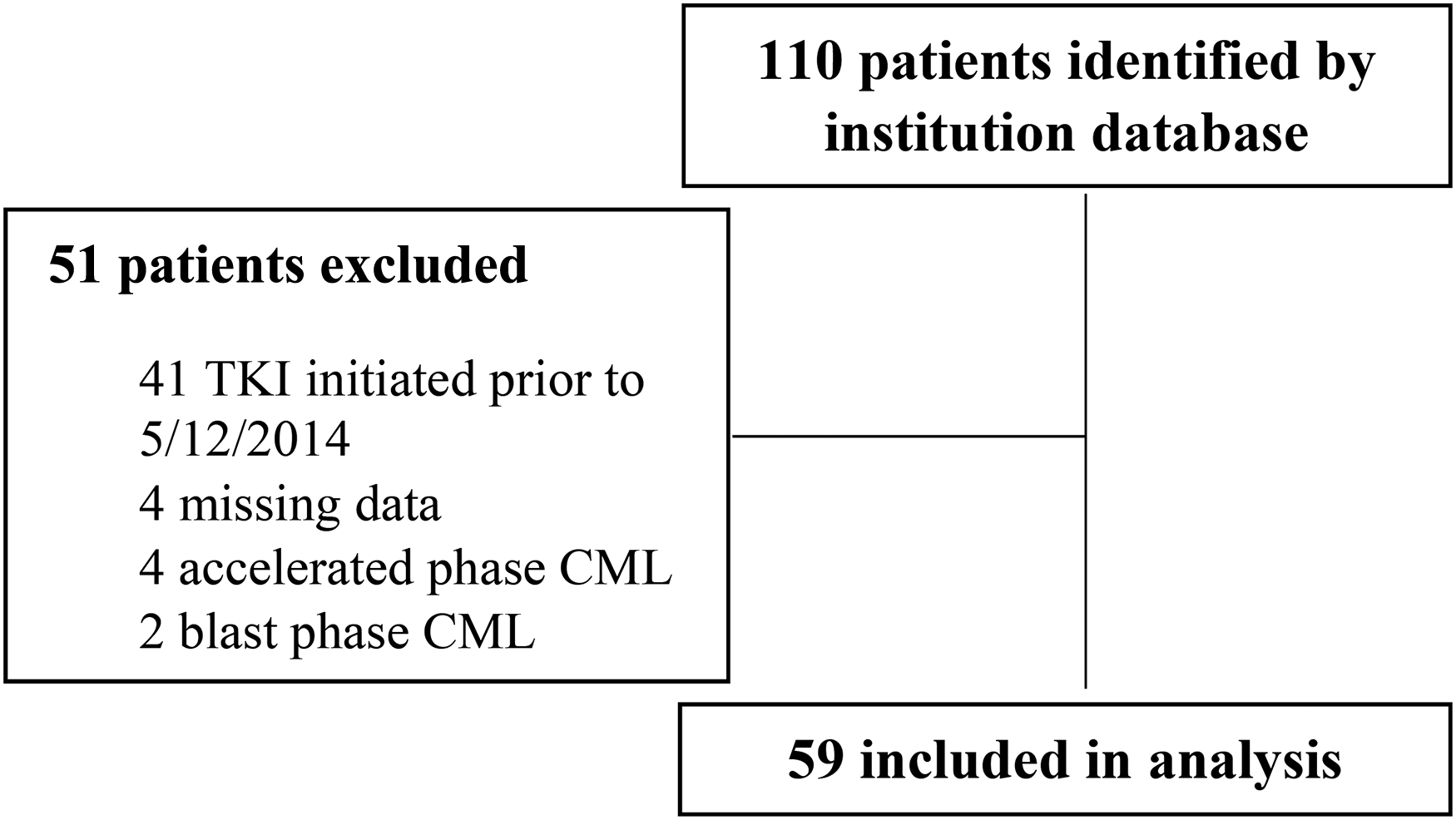
CONSORT diagram.
Baseline characteristics.
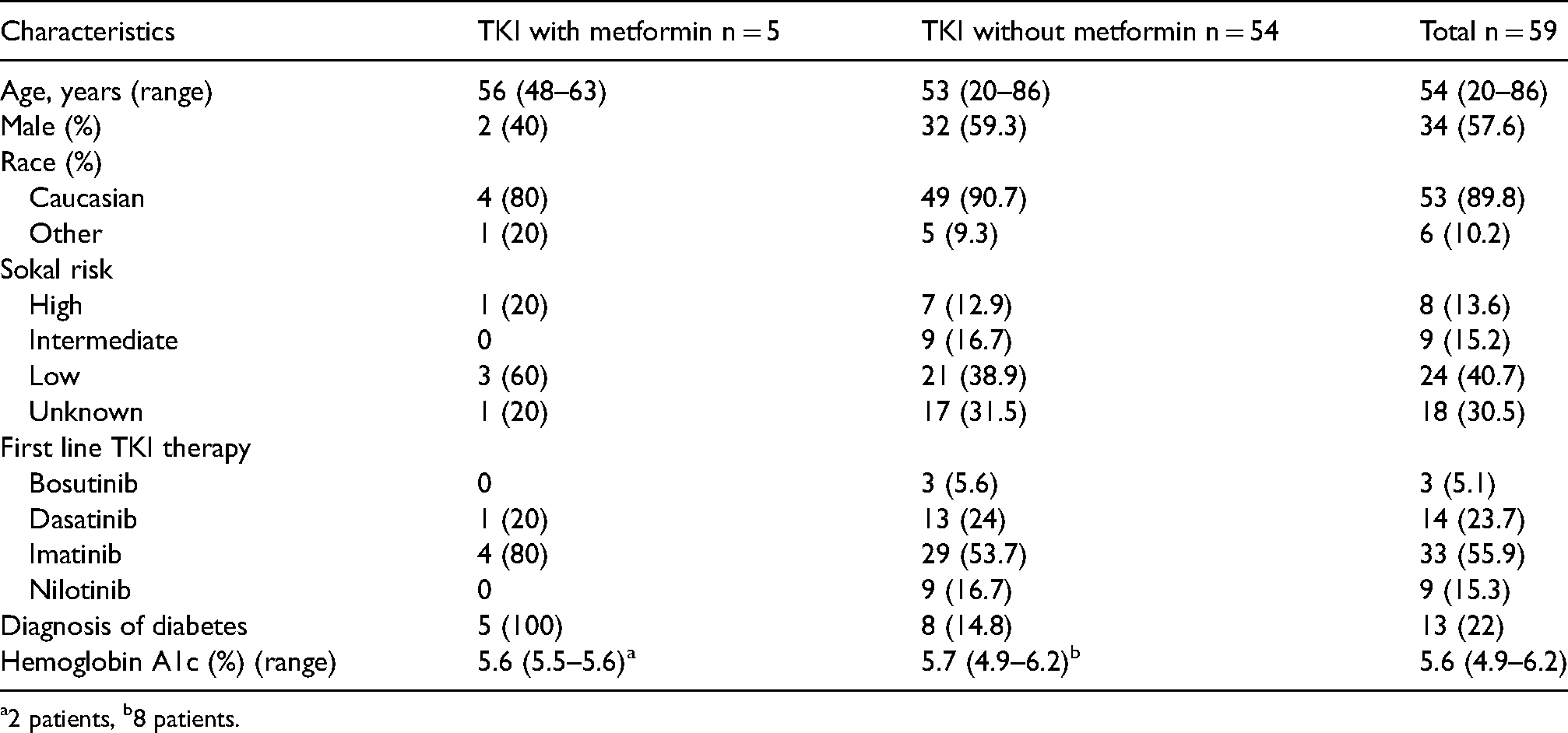
a2 patients, b8 patients.
Outcomes
Cytogenetic and molecular response results are shown in Table 2. The primary endpoint was the proportion of patients who achieved CCyR at one-year post-TKI initiation. One-hundred percent of patients in the metformin group achieved CCyR at one year compared to 74% in the non-metformin group (Table 2).
Primary and secondary endpoint results.
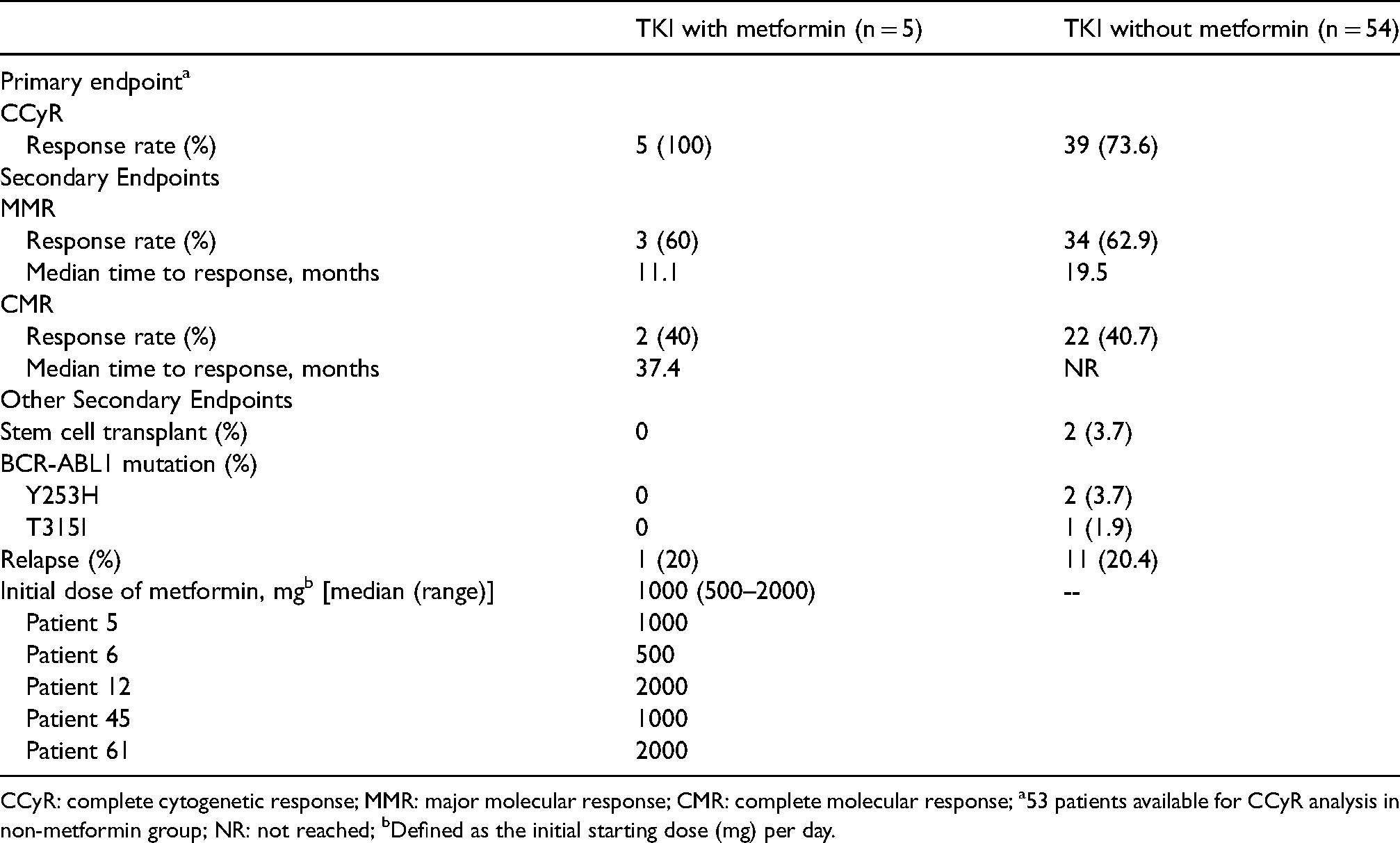
CCyR: complete cytogenetic response; MMR: major molecular response; CMR: complete molecular response; a53 patients available for CCyR analysis in non-metformin group; NR: not reached; bDefined as the initial starting dose (mg) per day.
Secondary endpoints are shown in Table 2. Approximately 60% of patients in both groups achieved MMR, with median time to MMR numerically shorter in the metformin group compared to the non-metformin group (11.1 vs. 19.5 mos). The proportion of patients achieving CMR was similar between both groups (approximately 40%), with median time to CMR 37.4 mos in the metformin group and not reached in the non-metformin group. Two patients taking a TKI without metformin received stem cell transplantation, whereas none of the 5 patients taking metformin received transplantation. Further, no patient taking a TKI plus metformin developed BCR-ABL1 mutation compared to three in the non-metformin group. Approximately 20% of patients in both groups had relapsed (loss of CCyR).
Discussion
Current initial standard of care for CML is TKI therapy. Patients achieving a deep molecular response and maintaining this for a minimum of 2 years on TKI therapy, may qualify for a TFR trial. However, not all who attempt to discontinue therapy are able to maintain their response. Because TKIs do not fully eliminate CML stem cells, TFR rates are imperfect (usually ∼40%–50% of patients maintain TFR).53–55 When TFR is unsuccessful, treatment re-initiation not only increases costs, but potentially decreases quality of life resulting from ongoing therapy-related adverse events.56–58 Adherence, at any point in time, may be hampered by adverse events leading to diminished dose intensity and lack, or loss of response. Ultimately, this unfortunately translates to diminished overall survival for some. Moreover, despite perfect adherence, some patients still fail to achieve CMR, which diminishes the chance that they may someday qualify for a TFR trial. Because suboptimal TFR rates could be substantially improved, the ability to eliminate CML stem cells remains a priority. Due to resistance of CML stem cells despite BCR-ABL1 inhibition, novel strategies are needed if we hope to eradicate CML stem cells, improve upon current TFR rates, and improve survival.
Several molecular pathways have been proposed as mediators of CML stem cell resistance and persistence in the face of continued TKI therapy. In recent years, a growing body of in vitro and murine literature has elucidated the potential antileukemic activity of antidiabetic agents. Peroxisome proliferator-activated receptor-gamma (PPAR-γ) agonists are a common class of medications prescribed for patients with diabetes. PPAR-γ agonists in combination with TKI therapy were initially thought to downregulate transcription of STAT5 and transition CML stem cells out of quiescence, thereby sensitizing them to TKI therapy.41,43–45 While Rousselot and colleagues reported that in CML patients on imatinib for a minimum of 2 years who already achieved MMR, the addition of pioglitazone increased deep molecular response rates. 45 More recent data revealed that pioglitazone did not appear to improve TFR trial results. 59 Although these data require confirmation within the constructs of a randomized clinical trial, they support PPAR-γ agonism and interference with cellular energy availability as a potential synergistic mediator of CML stem cell sensitization to TKI therapy.
Metformin, a more commonly used antidiabetic agent, has been extensively studied in solid tumor malignancies for its antineoplastic properties and has been shown in vitro to inhibit mTOR activity through 5’ adenosine monophosphate-activated protein kinase (AMPK)-dependent inhibitory effects. Of great interest owing to metformin's mechanism of action, accelerated phase and blast phase CML cells may express additional type 1 insulin-like growth factor receptors (IGF-1R) or increase IGF-1 ligand expression to upregulate BCR/ABL1 amplification through a phosphatidylinositol 3-kinase (PI3K)-, protein kinase B (AKT)-, mammalian target of rapamycin (mTOR)-dependent autocrine signaling mechanism.38,49–52,60 The proposed IGF-1R signaling cascade leading to stimulation of BCR-ABL1-independent growth signaling is shown in Figure 3. In this pathway, expression of IGF-1 receptors or upregulation of autocrine signaling by increased IGF-1 ligand expression may be inhibited by upregulation of AMPK or direct inhibition of IGF-1R by metformin. Additionally, increased IGF-1R signaling has also been shown as a potential TKI-resistance mechanism in non-BCR-ABL-directed TKI therapy. For example, crizotinib is an oral multitargeted TKI recommended for patients with non-small cell lung cancer (NSCLC) harboring specific tyrosine kinase rearrangements. In an in vitro model with crizotinib-resistant human lung cancer cells with increased IGF-1 signaling, metformin restored sensitivity by inhibiting this signaling pathway. 49 Other proposed molecular mechanisms for metformin's antileukemic activity and synergy with TKI therapy are illustrated in Figure 1 and have also previously been reported elsewhere.22–32,37,39 Based on myriad in vitro data across multiple tumor types, it is plausible that metformin possesses undiscovered antileukemic properties, such as those previously discussed.
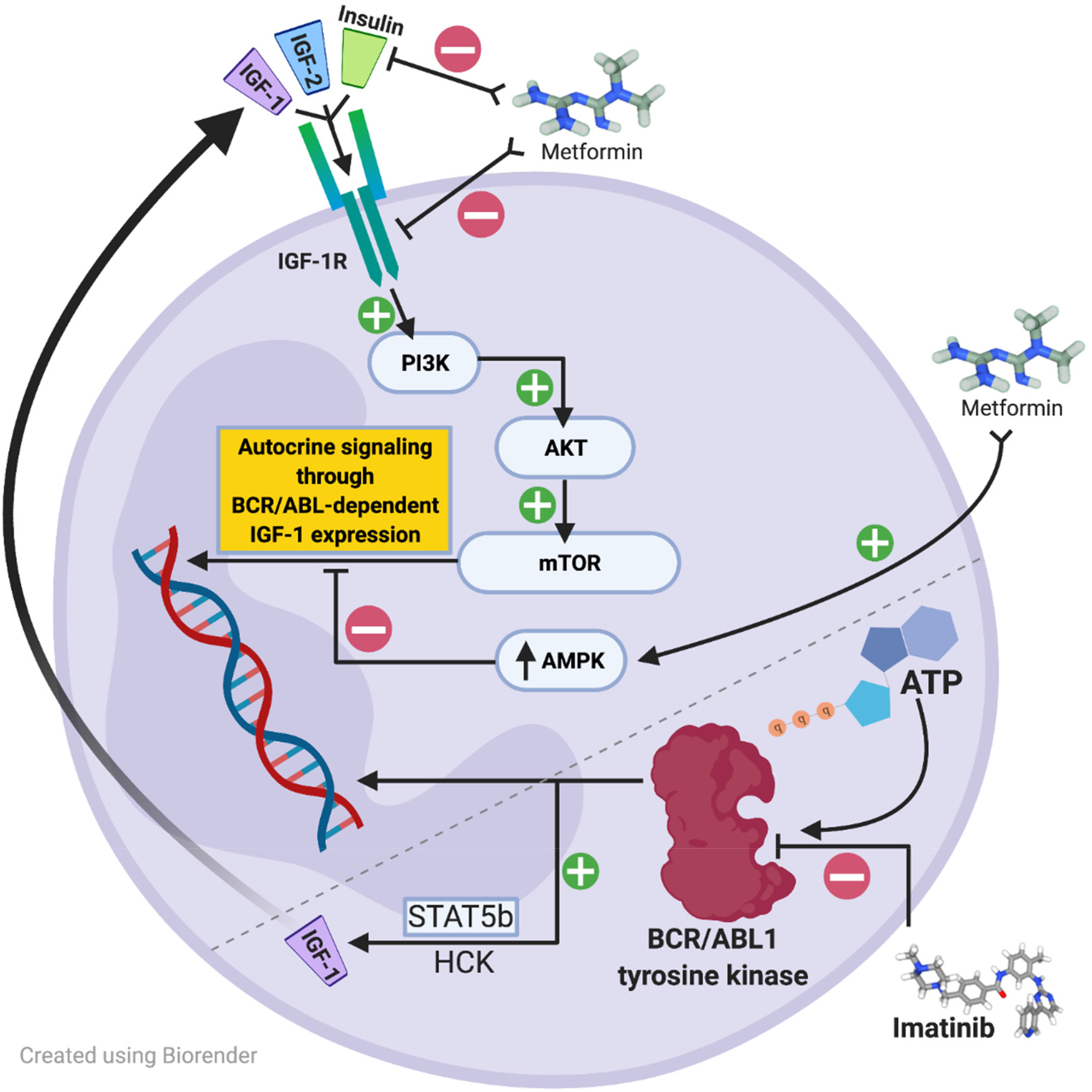
This observational study is the first to report on the use of metformin in combination with CML-directed TKI therapy. The percentage of patients achieving CCyR in the metformin group and the non-metformin group was 100% and 73.6%, respectively, with attainment of MMR and CMR numerically similar between both groups (Table 2). Median time to MMR and CMR were numerically shorter in the metformin group compared to the non-metformin group (11.1 mo vs. 19.5 mo; 37.4 mo vs. NR).
The main limitation of the current study was small sample size. A greater percentage of patients in the metformin group had low-risk disease, with 4 out of 5 (80%) initiated on a first generation TKI. Additionally, medication adherence and TKI dose interruptions/reductions were not fully assessed due to limitations inherent to an observational study design.
In conclusion, the results of this study suggest that metformin together with TKI therapy may increase the proportion of CML patients who achieve CCyR and decrease time to both MMR and CMR. The impact of metformin on TKI response rates warrants further investigation into whether metformin adds antileukemic synergistic properties. Confirmation of clinical benefit requires a large, prospective randomized controlled trial.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.