
Abstract
Purpose
Calcium levofolinate (CaLev) for intravenous administration is commercially available as a powder that must be reconstituted for injection or reconstituted and then diluted before administration. The lack of stability data on CaLev solutions renders necessary extemporaneous manual preparation, preventing the use of automated/semi-automated systems, with a consequent loss in terms of quality and safety.
Methods
The present work assessed the chemical–physical and microbiological stability of CaLev prepared in sodium chloride 0.9%, glucose 5% and water for injections and stored in polyolefin/polyamide bags and polypropylene syringes at 2–8°C protected from light. For this purpose, we developed and validated a new rapid High Performance Liquid Chromatography with Ultra Violet Diode-Array Detection (HPLC-UV-DAD) method.
Results
The samples tested were stable for 14 days, retaining >95% of their initial concentration and showing no change in colour, turbidity or pH. Microbiological tests performed on the samples were negative.
Conclusions
Our results confirmed the analytical stability of CaLev in NaCl 0.9%, glucose 5% and water for injection at concentrations used in clinical practice at our institute. This enables our centralized laboratory to organize the preparation of this drug in advance and the use of robots rather than manual preparation reduces the workload and the risk of preparation errors.
Introduction
Levofolinic acid, the L-diastereoisomer of the 5-formyl derivative of tetrahydrofolic acid, is the naturally occurring active form of the drug. The use of the active enantiomer reduces the doses administered to the patient by 50%, with no non-active drug for the body to.1,2 Calcium levofolinate (CaLev) has the same indication as folinic acid (FA) salts. It is used in cancer therapy to rescue normal cells from the toxic effects of high-dose antifolate therapy, such as methotrexate, 3 or to enhance the cytotoxic effects of 5-fluorouracil (5-FU) by stabilizing its binding to the thymidylate synthase enzyme. 4 For example, in the treatment of colorectal cancer, the combination 5-FU/FA compared to monotherapy with 5-FU leads to an improved response rate and overall survival. 5 The combination of 5-FU, FA and oxaliplatin or irinotecan also results in a significant increase in response, with an extension of progression-free survival (PFS) and overall survival (OS).6–8
The Oncology Pharmacy Unit of our institute (Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) IRCCS) comprises a centralized laboratory which is organized to prepare anticancer drugs up to 24 h before administration. Advance preparation has several benefits including a reduction in potential medication errors, better safety control, high assurance of stability, sterility and standardization of drug preparation.9–11 The laboratory is also equipped with robotic systems (APOTECAchemo®, Loccioni, Angeli di Rosora, Italy) that permit automated production and provide support technology for manual production.
APOTECAchemo® (Loccioni) is a robotic system that weighs active ingredients and solutions, reconstitutes powdered drugs and prepares syringes, bags and other final containers. The central part of the system, located within a closed and microbiologically controlled environment, comprises a robot with an anthropomorphic arm that mechanically replicates the manual actions of a human operator. The system permits us to continuously verify and check the entire preparation process. All of the production steps, as well as incoming and outgoing materials, are checked and registered by technological controls such as sensors, photocells, a vision system and barcode readers. The automatic identification of drugs, weight-checking and barcode labelling are used to guarantee complete traceability of the process. Robotic compounding improves patient and worker safety (closed working process) and increases workflow efficiency in terms of time optimization and efficient use of resources (both drugs and equipment). 12 Shifting from manual to robotic compounding is also a means of reducing the risks associated with drug preparation activities. 13
CaLev is not an anticancer drug as it does not have a cytotoxic mechanism of action. 14 Despite this, we decided to shift its preparation from manual to robotic compounding, normally reserved for anticancer drugs, because of its impact on the laboratory work-flow.
Currently marketed CaLev for intravenous administration is only available as a powder and must be reconstituted for injection or reconstituted and then diluted before administration. In accordance with several validated IRST therapeutic protocols, CaLev is administered in syringes after reconstitution alone or in bags with 5% glucose or 0.9% NaCl. The Summary of Product Characteristics sheet only reports data on the 12-h stability of the reconstituted drug protected from light 15 but does not provide any information on the stability of the diluted drug.
The lack of such stability data makes it impossible to dilute CaLev in advance to prepare infusion bags using automated/semi-automated systems (APOTECAchemo® and APOTECAps®, Loccioni). In fact, the robotic system allows only the preparation of the finished product, and after having reconstituted the drug, it must necessarily dilute it. Thus, the only option remaining is manual preparation, with a consequent reduction in quality and safety with respect to automated preparations.
To optimize the use of CaLev reconstituted in water for injections or reconstituted and diluted in sodium chloride (NaCl) or glucose, further data are needed on the stability of the drug up to at least 48 h stored at 2–8°C protected from light. To this purpose, we initially performed a search of the literature but found few indications that were useful for our specific setting. For example, Lebitasy et al. demonstrated the stability of CaLev diluted in 5% dextrose at a final concentration of 1.60 mg/mL in polyolefin bags frozen for 95 days at −20°C, microwave thawed according to a home-made validated ‘light cycle’ and stored for one month at 5 ± 3°C. 16 The data reported in the article were of little use to us as we prefer not to use freeze-thaw processes. Karbownik et al. showed that calcium folinate, either undiluted in glass containers or diluted with NaCl 0.9% in polyethylene bags, remained stable (<10% degradation) for at least 30 days at room and refrigerator temperatures when protected from light. 17 The data were not pertinent for our laboratory practice because APOTECAchemo robots cannot work with polyethylene bags, which are normally used for ancillary drugs. Our aim was to evaluate the stability of CaLev in bags that robots can work with.
Thus, given the paucity of useful information, we decided to conduct a new stability study on CaLev. The stability of a drug during storage is defined as the time during which its integrity in terms of quantity and chemical identity remains acceptable according to regulatory requirements. 18 A simple definition of stability based on a change in the concentration of active ingredients within a margin of ±10% is too simplistic. Most stability studies consider this range as a reference.Compared to considering a fixed arbitrary range, to define the maximum allowable concentration variation, a better approach requires that parameters such as therapeutic aspect, potential toxic risk of degradation products and possible instability of a preparation be considered.19,20 The procedures of our hospital require that drug prepared by the pharmacy should have a concentration that does not deviate by more than ±5% compared to that prescribed by the physician; this is the range what we consider for stability studies, to ensure maximum safety for our cancer patients and the maximum match between the prescribed dose of drug and that prepared, providing preparations with higher accuracy in the dosage. If a preparation has a dose variation greater than ±5%, the pharmacist can evaluate whether to repeat it or correct it.
Given that our Oncology Pharmacy Unit performs microbiological tests and has a High Performance Liquid Chromatography with Ultra Violet Diode-Array Detection (HPLC-UV-DAD) system that can be used to study the chemical stability of drugs, we decided to evaluate the stability of CaLev reconstituted in polypropylene syringes and diluted in glucose 5% and NaCl 0.9% in polyolefin/polyamide bags at concentrations for normal use stored for 14 days at 2–8°C protected from light.
Materials and methods
Additives, vehicles and materials
The following materials were used: CaLev TEVA 175 mg®; 0.9% sodium chloride injection, USP, 100 mL Viaflo Plastic Container (Baxter S.p.A., Rome, Italy); glucose 5% w/v intravenous infusion BP, 100 ml Viaflo Plastic Container (Baxter S.p.A., Rome, Italy); BACT/ALERT® (BioMerieux Italia S.p.A., Bagno a Ripoli, Italy); Acetonitrile-RS-for HPLC-PLUS-Gradient Grade (Carlo Erba Reagents S.a.s., Milan, Italy); Water HPLC Plus (Carlo Erba Reagents S.p.A., Milan, Italy); ammonium acetate (Carlo Erba Reagents S.a.s., Milan, Italy); hydrochloric acid (Carlo Erba Reagents S.a.s., Milan, Italy); sodium hydroxide (Carlo Erba Reagents S.a.s., Milan, Italy); hydrogen peroxide (Farmac-Zabban S.p.A, Bologna, Italy); and sodium hypochlorite (Lombarda H S.r.l, Milan, Italy).
Instrument and chromatographic conditions
Chromatographic analyses were performed using the Agilent 1200 HPLC system (Agilent Technologies, Santa Clara, California, USA) equipped with ChemStation revision B.04.01, Degasser G1379B, Binary Pump G1312A, Autosampler (ALS) G1329A, Thermostatted Column Compartment (TCC) G1316A and Diode-Array Detector (DAD) G1315D. Merck SeQuant® ZIC®-HILIC (3.5 µm, 200 Å) 150 × 4.6 mm column was used for separation. The column temperature was set at 25°C. The mobile phase that consisted of water and acetonitrile (40/60, v/v) adjusted to pH 4.8 with ammonium acetate buffer (20 mM) was used for isocratic elution at a flow rate of 1 ml/min. The injection volume was 1 μl. The eluent was monitored at 267 nm for drug detection over a period of 7 min. The pH measurement of the CaLev was performed with the pH meter XS Instruments pH 7+ DHS.
Preparation of standard and sample solutions
Preparation of standard solutions for validation process
All samples were set up using Gilson® pipettes in Agilent glass vials supplied with the HPLC system in use and suitable for the autosampler. The different concentrations of the analysed samples were prepared by diluting with water for HPLC the drug (10 mg/mL) taken from the reconstituted vials with water for injections. After mixing and agitating with the vortex mixer, the sample was visually inspected to verify the absence of precipitates, transparency and correct sample setup (volume matching by comparison with the graduated scale on glass vials). Each sample was then filtered further using special 0.45 µm KX syringe filters. Each sample was injected into the column three times. The drug itself was used as standard for the construction of the calibration curve in that, not having excipients that absorb at the same wavelength as the drug or that interact with the drug, it was considered pure. For the validation protocol, samples were used at concentrations of 0.175 mg/mL (C1) and 0.7 mg/mL (C2).
Preparation of CaLev infusion
The doses of CaLev assayed in the study were chosen on the basis of the doses most frequently used in our clinical practice, i.e.10 mg/mL for the syringes and 1.7 mg/mL for the bags. For injections, the CaLev powder was reconstituted with 17.5 mL of water, obtaining a product with a concentration of 10 mg/mL of CaLev, ready for use with syringes. For the bags, 17.5 mL of solution were removed from each bag (glucose and sodium chloride) and replaced with 17.5 mL of reconstituted CaLev. Due to the overfilling of the bags with the diluent, the final concentration was 1.68 mg/mL instead of 1.75 mg/mL.
The following quantities were prepared:
three 17.5 mL syringes with water for injection; three 100 mL bags with 0.9% sodium chloride for injection; three 100 mL bags with 5% glucose for injection.
The drugs were prepared under aseptic conditions in laminar flow hoods and stored at 2–8°C well protected from light in yellow polythene overwraps.
Validation of the HPLC method
After the analytical conditions had been optimized, linearity, precision, accuracy (recovery), selectivity, limit of detection (LOD), limit of quantification (LOQ) and robustness were evaluated according to the International Council for Harmonisation (ICH) of Technical Requirements for Pharmaceuticals for Human Use guidelines. 21
System suitability
In accordance with United States Pharmacopeia (USP) criteria, 22 system suitability was checked by determining the peak retention time and the area, resolution, selectivity factor, theoretical plates and symmetry factor for CaLev. The sample was prepared as previously described (see Preparation of standard solutions for validation process section). Nine replicate samples were assayed to determine the system suitability.
Specificity
A C2 sample and water sample for HPLC were used to evaluate the specificity of the method. Samples were prepared as previously described (Preparation of standard solutions for validation process section). Acceptance criteria: The interference of the diluent was considered non-significant if its chromatogram showed no peaks at the retention time of CaLev. 23 The excipients in the drug are mannitol, sodium hydroxide and hydrochloric acid; these do not interfere with the analysis of CaLev, since they do not absorb at the wavelength in which the analyses were performed.
Range and linearity
To establish linearity, a calibration line was constructed from plots of peak areas vs. concentrations using eight different samples at different concentrations (from 0.05 mg/mL to 1.3 mg/mL) and each sample was analysed three times (Preparation of standard solutions for validation process section). Acceptance criteria: correlation coefficient, R2 ≥ 0.99.
Detection limit and quantitation limit
The LOD and LOQ were determined using the standard deviation of the response and the slope.
LOD = 3.3σ/S LOQ = 10σ/S
where σ is the standard deviation of the response and S is the slope of the calibration curve.
Accuracy/recovery
The accuracy of the method was assessed as its ability to quantify the concentration of C1 and C2 samples (Preparation of standard solutions for validation process section) by comparing the theoretical concentration of the samples with that of the experimental concentration. Three samples were prepared for each concentration, and each was analysed three times. All of the analyses were carried out on the same equipment, on the same day, by the same analyst.
Precision
Precision was evaluated by determining the repeatability and intra-day and inter-day variability.
Repeatability refers to the degree of agreement between several independent measures of the same analytical variable. To estimate repeatability, standard deviation values and coefficients of variation of the C1 and C2 samples were determined (Preparation of standard solutions for validation process section). One sample was prepared for each concentration, and each sample was analysed nine times. All of the analyses were carried out on the same equipment, on the same day, by the same analyst.
Intra-day variability was evaluated by performing two sets of analyses at a distance of 12 h from each other on three samples at C1 and C2 concentrations (Preparation of standard solutions for validation process section). Each sample was analysed three times. All of the analyses were carried out on the same equipment by the same analyst.
Inter-day variability was assessed by analysing the standard deviation and the coefficient of variation of the analyses conducted on C1 and C2 samples on days 0, 3 and 7. Three samples were prepared each of these days, and each was injected three times.
Stability indication
The stability indicating capability of the method was evaluated by determining its ability to distinguish the drug from its degradation products.16,19,24 The degradation products were obtained by treating 1 mL of drug (0.7 mg/mL) with 1 µL of 2.7% sodium hypochlorite for 20 min, 50 µL of 3% hydrogen peroxide for 20 min and 1 mL of sodium hydroxide 0.2 N for 12 h.
Robustness
The robustness of the method was evaluated by deliberate variation of chromatographic parameters such as column temperature, mobile phase composition, flow rate and mobile phase pH. Three samples were prepared at C2 concentration (Preparation of standard solutions for validation process section) and each one was analysed three times (nine measurements/point). The mean retention time (Tr’) and mean peak area were determined for CaLev against each setting. In all cases, the effects of small changes made to the method were determined by evaluating the percentage variations of the values obtained, with respect to the mean retention time and the mean area obtained in the standard conditions of the method.
Stability studies
Physical stability
The physical stability of the samples was checked visually during the study period for changes in colour, turbidity and/or precipitation according to the European Pharmacopoeia 9th edition. 25 An exhaustive control of the pH variation as a function of the time was also carried out, considering the modification of 1 unit of the pH value as the maximum acceptable variation.
Chemical stability
The chemical stability of mixtures was evaluated by determining the amounts of drugs present in each device and evaluating the absence of degradation products by HPLC. For this purpose, samples were taken from the sodium chloride 0.9% and glucose 5% bags using a syringe without a needle inserted through the spike on the bags, and from syringes containing CaLev. The samples taken from the bag were immediately diluted with water for HPLC to obtain a concentration of 0.8 mg/mL, while the syringe samples were diluted to a concentration of 0.7 mg/mL. Three samples were taken from each bag and syringe and each sample was analysed three times. The samples were analysed immediately after preparation (t = 0) and at the following scheduled time intervals: 1, 2, 3, 6, 9, 12 and 14 days (see Preparation of CaLev infusion section).
Microbiological stability
Microorganisms and enzymatic systems can pollute preparations and give rise to different degradation reactions (oxidation, reduction and hydrolysis). These reactions can alter the organoleptic properties of the drug leading to a loss of its activity, in addition to posing a risk for the patient due to the presence of pathogenic germs.
For the microbiological analysis we analysed two samples for each type of preparation using BACT/ALERT® culture bottles which are a simple, automated rapid microbial detection system capable of detecting bacterial, yeast and mould contamination in a wide variety of matrices (food, beverage, drugs, etc.).19,26–28 Each BACT/ALERT® bottle contains sterile culture medium and is imbedded with a colorimetric sensor that changes from grey to yellow in the presence of CO2 produced by growing microorganisms. Once bottles were loaded, the colorimetric sensors were scanned every 10 min. If growth was detected, the system set off both audible and visual alarms and the sample data were recorded. In the present study, the microbiological was carried out 14 days after drug preparations.
Results
Validation of the HPLC method
The HPLC-UV-DAD method used in our study was fully validated, as reported in the Materials and methods section. The parameters of system suitability are summarized in Table 1. All measured parameters were within the recommended limits according to the USP. Thus, our results suggested that the described method is suitable for the determination of CaLev with a retention time of about 2.1 min. A typical chromatogram of CaLev and its UV spectrum are shown in Figure 1(a).
Statistical analysis of parameters.
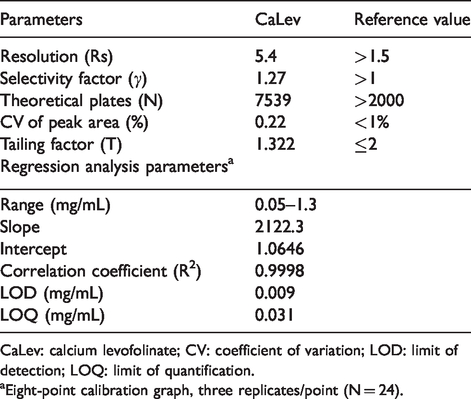
CaLev: calcium levofolinate; CV: coefficient of variation; LOD: limit of detection; LOQ: limit of quantification.
aEight-point calibration graph, three replicates/point (N = 24).
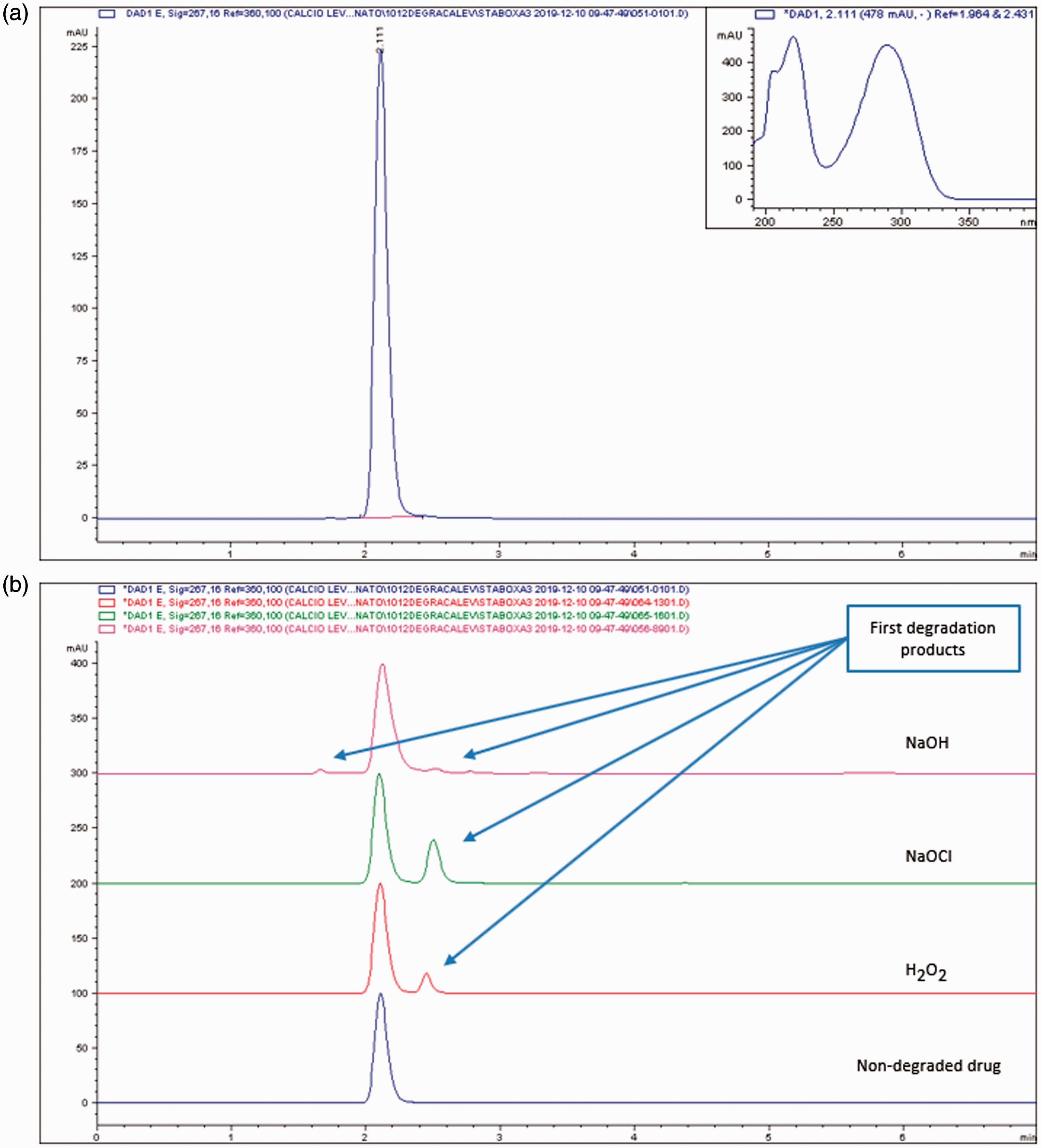
(a) Chromatogram of CaLev (0.7 mg/mL) with a retention time of 2.111 min detected at 267 nm. (b) CaLev non-degraded (blue curve), treated with 3% hydrogen peroxide (H2O2) red curve, 2.7% sodium hypochlorite (NaOCl) green curve and sodium hydroxide 0.2 N (NaOH) violet curve.
The specificity of the method was achieved using the UV-DAD detector which clearly showed the absence of interfering peaks in the diluent at the CaLev retention time. Good linearity was demonstrated by the high value of the correlation coefficient and the low intercept value (Table 1). The accuracy for CaLev at a concentration range of 0.175–0.7 mg/mL varied from 97.2% to 105.1%. The method was considered repeatable because the coefficient of variation percentage (CV%) for each sample analysed (C1 and C2) was <0.7. Intra-day variability data showed a non-significant difference in Tr’ values and concentrations of samples analysed at a distance of 12 h. Inter-day variability data revealed a non-significant difference in Tr’ values and concentrations of the analysed samples on days 1, 4 and 7. The total CV% of the analyses conducted was <2.5%, indicating that the developed method was precise.
The forced degradation analysis demonstrated the ability of the method to separate the intact drug from its first degradation products (Figure 1(b)).
The slight variations in column temperature, mobile phase composition, flow rate and pH did not lead to a significant difference in the retention time and peak area of the analytes. Thus, the developed method can be considered robust. For more information, see Supplementary Table A1.
Stability of CaLev solution for injection
All the samples, bags and syringes prepared as described in the Materials and methods section were analysed to assess physical and chemical stability. Variations in colour, precipitate formation and turbidity formation were monitored over a period of 14 days (2–8°C and light protection conditions). The average pH values of the drug stored in syringes, bags with 5% glucose solution and bags with 0.9% sodium chloride, from the day of preparation (day 0) to the 14th day, ranged from 7.95 to 7.44, 6.52 to 6.53 and 6.63 to 6.34, respectively. These changes did not affect the chromatographic parameters (retention time and peak area). During storage, variations in drug concentrations were <5% (Table 2) and no degradation products were observed. All the preparations subjected to microbiological testing were negative for growth of both aerobic and anaerobic microorganisms, confirming that our Oncology Laboratory was able to maintain aseptic conditions during the whole drug production process.
Stability data.
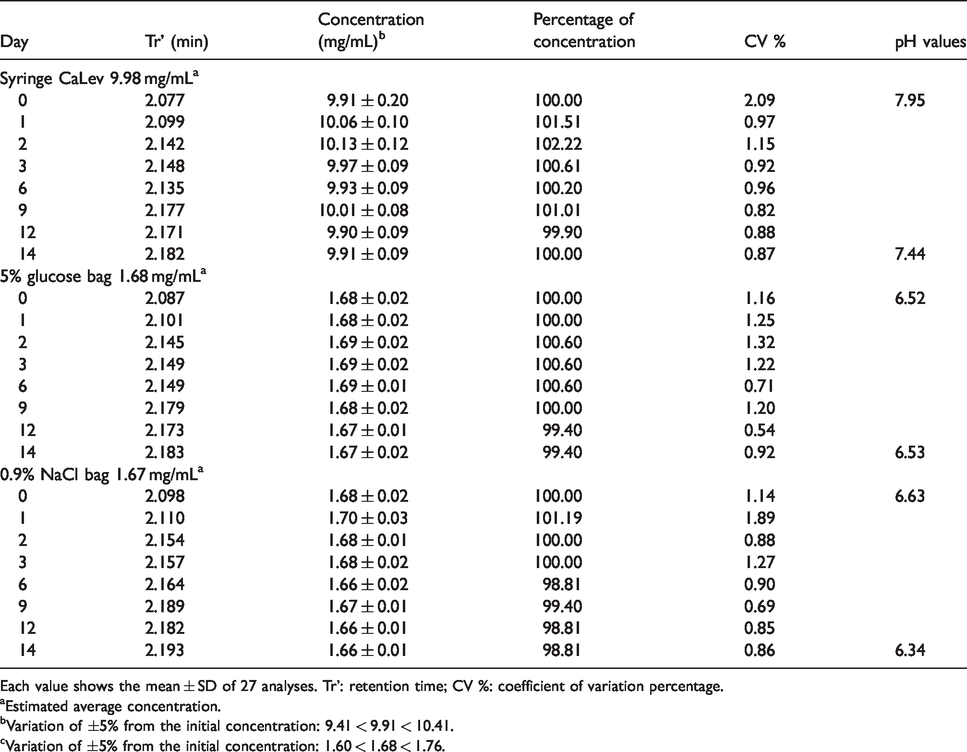
Each value shows the mean ± SD of 27 analyses. Tr’: retention time; CV %: coefficient of variation percentage.
aEstimated average concentration.
bVariation of ±5% from the initial concentration: 9.41 < 9.91 < 10.41.
cVariation of ±5% from the initial concentration: 1.60 < 1.68 < 1.76.
Discussion
The proposed RP-HPLC method for assaying CaLev was simple, precise, highly accurate and time-efficient. The intra-day and inter-day variability and accuracy results were also acceptable. The method was validated as per ICH Guidelines and stability studies under different conditions confirmed that it was highly robust, indicating its potential for routine use in research institutes. We also utilized the method to study the stability of CaLev at the usual concentration levels used in clinical practice.
Our results showed that CaLev prepared in 0.9% sodium chloride or in 5% glucose solution or in water for injection, stored at 2–8°C under light protection for 14 days is:
Chemically stable since the maximum variation in concentration recorded with respect to the initial one was +2.22%. Furthermore, no degradation products were observed in chromatograms and no variation of the UV absorption spectrum of the drug was recorded. Physically stable as no change in visual appearance was observed and the maximum measured pH change (0.51 pH unit) meets the proposed acceptance criteria of the ‘Methodological guidelines for stability studies of hospital pharmaceutical preparations’.
19
Microbiologically stable as during storage there was no growth of microorganisms in the preparations made under the laminar flow hoods. Since the microbiological stability is closely linked to the working methods of each production unit, once we obtained the chemical–physical stability data of the CaLev prepared under the laminar flow hood, we were able to transfer these preparations to the robot. This allowed us to repeat the microbiological tests also on the preparations made by the robot and these also tested negative after a storage period of 14 days. However, these results cannot be extended to other departments, since as discussed earlier, it depends on the working methods.
Conclusions
The results obtained allowed us to extend the stability data of CaLev to 14 days. These data will allow us to organize the preparations of drugs in advance using robots, avoiding extemporaneous manual preparations, reducing the workload and potential preparation errors and increasing the traceability and safety of the process.29,30
Furthermore, the automation of the production of CaLev has also had important repercussions in terms of the optimization of the robot technology in use in our institute. In 2017, around 1800 CaLev preparations were made manually in the IRST pharmacy. The increased use of robots has enabled us to reach a breakeven point, which represents the number of preparations beyond which the robotic production becomes advantageous compared to the manual production in terms of costs. 12
Supplemental Material
sj-pdf-1-opp-10.1177_1078155220918025 - Supplemental material for Stability of calcium levofolinate reconstituted in syringes and diluted in NaCl 0.9% and glucose 5% polyolefin/polyamide infusion bags
Supplemental material, sj-pdf-1-opp-10.1177_1078155220918025 for Stability of calcium levofolinate reconstituted in syringes and diluted in NaCl 0.9% and glucose 5% polyolefin/polyamide infusion bags by Seydou Sanogo, Paolo Silimbani, Raffaella Gaggeri and Carla Masini in Journal of Oncology Pharmacy Practice
Supplemental Material
sj-pdf-2-opp-10.1177_1078155220918025 - Supplemental material for Stability of calcium levofolinate reconstituted in syringes and diluted in NaCl 0.9% and glucose 5% polyolefin/polyamide infusion bags
Supplemental material, sj-pdf-2-opp-10.1177_1078155220918025 for Stability of calcium levofolinate reconstituted in syringes and diluted in NaCl 0.9% and glucose 5% polyolefin/polyamide infusion bags by Seydou Sanogo, Paolo Silimbani, Raffaella Gaggeri and Carla Masini in Journal of Oncology Pharmacy Practice
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.