
Abstract
In nucleated cells, the extrinsic pathway of the programmed cell death (apoptosis) is triggered by interaction of death ligands of the tumor necrosis factor superfamily with the death receptors on external cell surface membrane. In this review, we present evidence that, in contrast to nucleated cells, apoptosis in anucleate platelets can be induced through bypassing the death receptors, using instead specific receptors on the platelet surface mediating platelet activation, aggregation, and blood coagulation. These platelet surface receptors include the protease-activated receptor 1 of thrombin and glycoproteins IIbIIIa and Ibα, receptors of fibrinogen, and von Willebrand factor. The pro-apoptotic BH3 mimetic ABT-737 and calcium ionophore A23187 also trigger platelet apoptosis without using death receptors. These agents induce the intrinsic pathway of platelet apoptosis by direct targeting mitochondrial and extra-mitochondrial apoptotic responses.
Keywords
Introduction
Two pathways lead to apoptosis in nucleated cells. The extrinsic pathway is triggered by the interaction of death ligands belonging to the tumor necrosis factor superfamily with their cognate death receptors on the external cell surface membrane. After recruiting adapter proteins, this pathway results in the formation of active caspase-8, which serves as initiator caspase of the extrinsic pathway and activates the apoptosis executioners, caspases-3, -6, -7, which cleave vital cell proteins and shift the apoptotic process to the execution phase of apoptosis. 1 –9
The intrinsic pathway, on the other hand, is triggered by changes in mitochondrial integrity, which include depolarization of mitochondrial inner membrane potential (ΔΨm) and release of pro-apoptotic proteins, such as cytochrome c, from the mitochondrial intermembrane space, under the control of pro-apoptotic and anti-apoptotic proteins of Bcl-2 family. Once released from mitochondria, cytochrome c works together with cytosolic proteins – apoptotic protease-activating factor-1 and activated upstream caspase-9, which in turn activates executioner caspases-3, -6, -7. Thus, in nucleated cells, both extrinsic and intrinsic apoptosis pathways, separately or together, may shift an apoptotic process to the point of no return. 9 –17
In this report, we present data on triggering apoptosis in anucleate platelets, bypassing the death receptors of the extrinsic apoptosis pathway, using instead platelet surface receptors traditionally considered as receptors for platelet activation and aggregation and blood coagulation, including (1) the protease-activated receptor-1 (PAR-1) of thrombin, (2) glycoprotein IIbIIIa (αIIbβ3 integrin), receptor of fibrinogen and von Willebrand factor (VWF), and (3) glycoprotein GPIbα, another receptor of VWF.
Platelet Apoptosis Mediated by Protease-Activated Receptor-1 (PAR-1) of Thrombin
Currently, there is no evidence that death ligand-death receptor interactions can induce platelet apoptosis (PL-Apo) via the extrinsic apoptosis pathway. 18,19 Platelet apoptosis can however be triggered by the natural platelet agonist thrombin, 20 –25 a potent inducer of platelet activation and of converting fibrinogen to fibrin generating blood clotting. 26,27
As shown in Table 1, thrombin triggers PL-Apo, impacting 3 types of platelet responses: (1) mitochondrial (or mitochondria-associated) events – ΔΨm depolarization, shifting the balance between Bcl-2 regulatory proteins in a pro-apoptotic direction enhancing expression of Bax and Bak, and inducing activation and translocation to mitochondria of active Bid, Bax, and Bak; (2) extra-mitochondrial events – caspase-3 activation and phosphatidylserine (PS) exposure; and (3) cellular event – microparticle (MP) formation.
Execution of Platelet Apoptosis (PL-Apo) via Bypassing the Death Receptors: Mitochondrial, Extra-Mitochondrial, and Cellular PL-Apo Responses are Strongly Dependent on the Type of Specific Trigger and Mediator of PL-Apo.
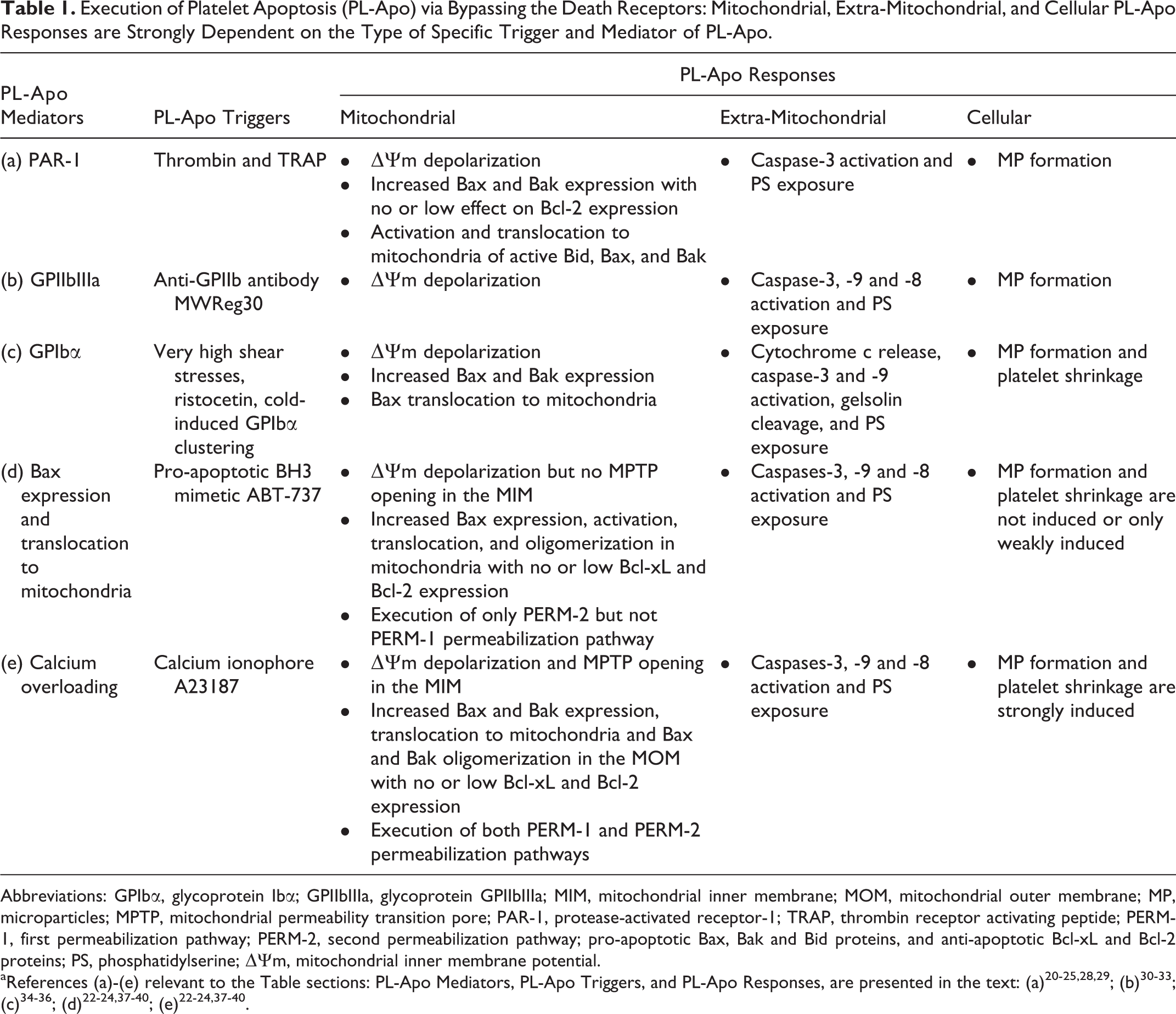
Abbreviations: GPIbα, glycoprotein Ibα; GPIIbIIIa, glycoprotein GPIIbIIIa; MIM, mitochondrial inner membrane; MOM, mitochondrial outer membrane; MP, microparticles; MPTP, mitochondrial permeability transition pore; PAR-1, protease-activated receptor-1; TRAP, thrombin receptor activating peptide; PERM-1, first permeabilization pathway; PERM-2, second permeabilization pathway; pro-apoptotic Bax, Bak and Bid proteins, and anti-apoptotic Bcl-xL and Bcl-2 proteins; PS, phosphatidylserine; ▵Ψm, mitochondrial inner membrane potential.
The ability of thrombin to induce PL-Apo suggests that, as in nucleated cells, 28,29 thrombin can trigger PL-Apo by cleavage of PAR-1. Treatment of platelets with the PAR-1 peptide agonist – thrombin receptor activating peptide (TRAP), induces translocation of pro-apoptotic Bid and Bax to mitochondria. 25 Thus, aside from its main functions as coagulation factor and inducer of platelet activation, thrombin triggers PL-Apo without participation of death receptors, using instead the PAR-1 platelet surface receptor. However, although thrombin at a high dose of 1 U/mL significantly induces key markers of PL-Apo, 20 these effects are relatively weak compared to the effect on platelet activation. 21 –24
Platelet Apoptosis Mediated by Glycoprotein IIbIIIa (GPIIbIIIa)
Platelet surface receptor GPIIbIIIa plays a key role in platelet aggregation, hemostasis, and thrombosis. 30,31 However, as shown in Table 1, data from a murine model of immune thrombocytopenia demonstrate that GPIIbIIIa may be directly involved in PL-Apo, since strong thrombocytopenia induced by injection into mice of anti-GPIIb antibody, resulting in a reduction of platelet count by approximately 75%, was associated with increase in ΔΨm depolarization, caspase-3 activation, and PS exposure. 32 Furthermore, clinical studies have shown that pediatric acute immune thrombocytopenia (often associated with GPIIbIIIa autoantibodies) was also associated with increased ΔΨm depolarization, caspase-3, -9, -8 activation, PS exposure, and MP formation. 33
Platelet Apoptosis Mediated by Glycoprotein Ibα (GP1bα)
As shown in Table 1, PL-Apo can be triggered by interaction of VWF with GPIbα receptor when this interaction is induced by: (1) biomechanical forces – very high shear stresses, indicating that GPIbα may function as mechanoreceptor transmitting apoptotic signals inside the platelet 34 ; (2) a chemical stimulus ristocetin, which requires interaction of intracellular signaling protein 14-3-3ζ with the cytoplasmic domain of GPIbα for GPIbα–VWF-induced PL-Apo 35 ; and (3) cold-induced GPIbα clustering which, after interaction with VWF, enhances PL-Apo signaling. 36
Platelet Apoptosis Triggered by the Pro-Apoptotic BH3 Mimetic ABT-737 and by Calcium Ionophore A23187
Table 1 shows that PL-Apo can be also triggered by the pro-apoptotic BH3 mimetic ABT-737 and by calcium ionophore A23187 and mediated by Bax expression and translocation to mitochondria and by calcium overloading, respectively. Both agents induce PL-Apo without using death receptors. Platelet apoptotic responses induced by ABT-737 are characterized by a wide spectrum of mitochondrial events, including (1) increased ΔΨm depolarization but with no mitochondrial permeability transition pore (MPTP) opening in the MIM; (2) increased Bax expression, activation, translocation, and oligomerization in mitochondria with no or low Bcl-xL and Bcl-2 expression; and (3) execution of only the second mitochondrial permeabilization pathway (PERM-2) but not the first permeabilization pathway (PERM-1). Extra-mitochondrial events include increased caspases-3, -9, -8 activation and PS exposure. However, ABT-737 is not able to induce, or only weakly induces, cellular apoptotic events such as MP formation and platelet shrinkage. 22 –24,37 –40
Table 1 summarizes data on PL-Apo triggered by A23187 and mediated by calcium overloading. Comparison between mitochondrial and cellular PL-Apo responses in A23187-treated platelets versus responses in ABT-737-treated platelets indicates that A23187 is a much stronger inducer of PL-Apo than ABT-737. 22 –24,37 –40 In mitochondrial responses, A23187-treated platelets are characterized by (1) increased ΔΨm depolarization and MPTP opening in the MIM, (2) increased Bax and Bak expression, translocation to mitochondria, and Bax and Bak oligomerization in the mitochondrial outer membrane with no or low Bcl-xL and Bcl-2 expression, and (3) execution of both PERM-1 and PERM-2 permeabilization pathways. In contrast, ABT-737-treated platelets are characterized by (1) no MPTP opening in the MIM, (2) only Bax expression followed by activation, translocation, and oligomerization in mitochondria with no or low Bcl-xL and Bcl-2 expression, and (3) execution of PERM-2 but not PERM-1 pathway of permeabilization. Comparison of the cellular responses, MP formation and platelet shrinkage, shows that A23187 strongly induces and ABT-737 does not induce or only weakly induces these responses.
Conclusions
Despite the absence of death receptors and the extrinsic pathway of apoptosis as in nucleated cells, anucleate platelets successfully perform apoptosis by using the mitochondrial intrinsic apoptosis pathway via PAR-1, GPIIbIIIa, or GPIbα platelet surface receptors. Thus, in addition to their main function of participation in platelet activation, aggregation and blood coagulation, PAR-1, GPIIbIIIa, and GPIbα receptors can be involved in PL-Apo. The pro-apoptotic BH3 mimetic ABT-737 and calcium ionophore A23187 can also trigger PL-Apo bypassing the death receptors, directly affecting the intrinsic pathway of apoptosis by inducing mitochondrial and extra-mitochondrial manifestations of PL-Apo.
Footnotes
Authors’ Note
V.L., A.V.G., and J.F. wrote the paper.
Acknowledgments
The authors thank D. J. Allen, S. Mykhaylov, A. Mutlu, J. W. Semple, A. H. Lazarus, E. Lyubimov, H. Ni, and B. Garvey for research cooperation.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from the Platelet Research Fund of Ronya Beskin, Israel.