
Abstract
Present review highlights some new aspects of the role of individual components of blood coagulation process and proposes a modified concept of hemocoagulation cascade. The role of FXII in the initiation of the so-called intrinsic coagulation system is currently questioned. Its role has been recently demonstrated mainly in the thrombus propagation and final stabilization together with factors XI and XIII. The edited concept underlines the common part of the tissue factor (TF) in the initiation of both the intrinsic and extrinsic pathways of the coagulation system and therefore may make it not improperly be called the TF coagulation pathway. The search for new antithrombotic agents shows that the level of the coagulation system blockade depends on which step in the coagulation cascade is targeted and results in different degrees of the antithrombotic efficiency and the risk of bleeding complications.
Introduction
Recent advances have shifted significantly our understanding of the process of coagulation, both in cardiovascular disease and in the wider context of coagulation as a whole. Experimental work, the development of new antithrombotic agents as well as a new look at older dogmas, has gradually and fundamentally changed our understanding of the processes of coagulation.
Arterial or venous thrombosis is responsible for more than half of all deaths worldwide, and recently, we have witnessed the production and clinical use of new antithrombotic agents, and their efficient development continues. Broad introduction and clinical usage of new antithrombotic drugs led to the promotion of knowledge about thrombosis and antithrombotic treatment. Basic understanding of pathophysiological mechanisms of coagulation, however, recently went much further, and it is essential for the proper management of antithrombotic therapy.
Isolation of blood from other tissues to ensure its laminar circulation is maintained by series of interrelated mechanisms, and dynamic equilibrium between blood circulation and reparation processes has a delicate balance in a very complex system of blood coagulation, spontaneous anticoagulation, and fibrinolysis.
These processes are studied for many decades by clinical description and laboratory examination of hemocoagulation of patients with deficiency of procoagulant and anticoagulant factors (F; enzymes and proteins), in experiments on animals, by in vitro assays, and recently, also by mathematical and computer models as well as by using specific blocking antibodies of either individual factors or by reverse blockers of such antibodies themselves.
The Edited Concept of Coagulation
Progress of coagulation can be generally divided into the following stages (Figure 1): initiation, amplification, propagation, and stabilization. The most significant changes, in this coagulation scheme, are relating to the existence of both the extrinsic and intrinsic pathways of the coagulation system, the role of tissue factor (TF), and FXI, FXII, and FXIII.
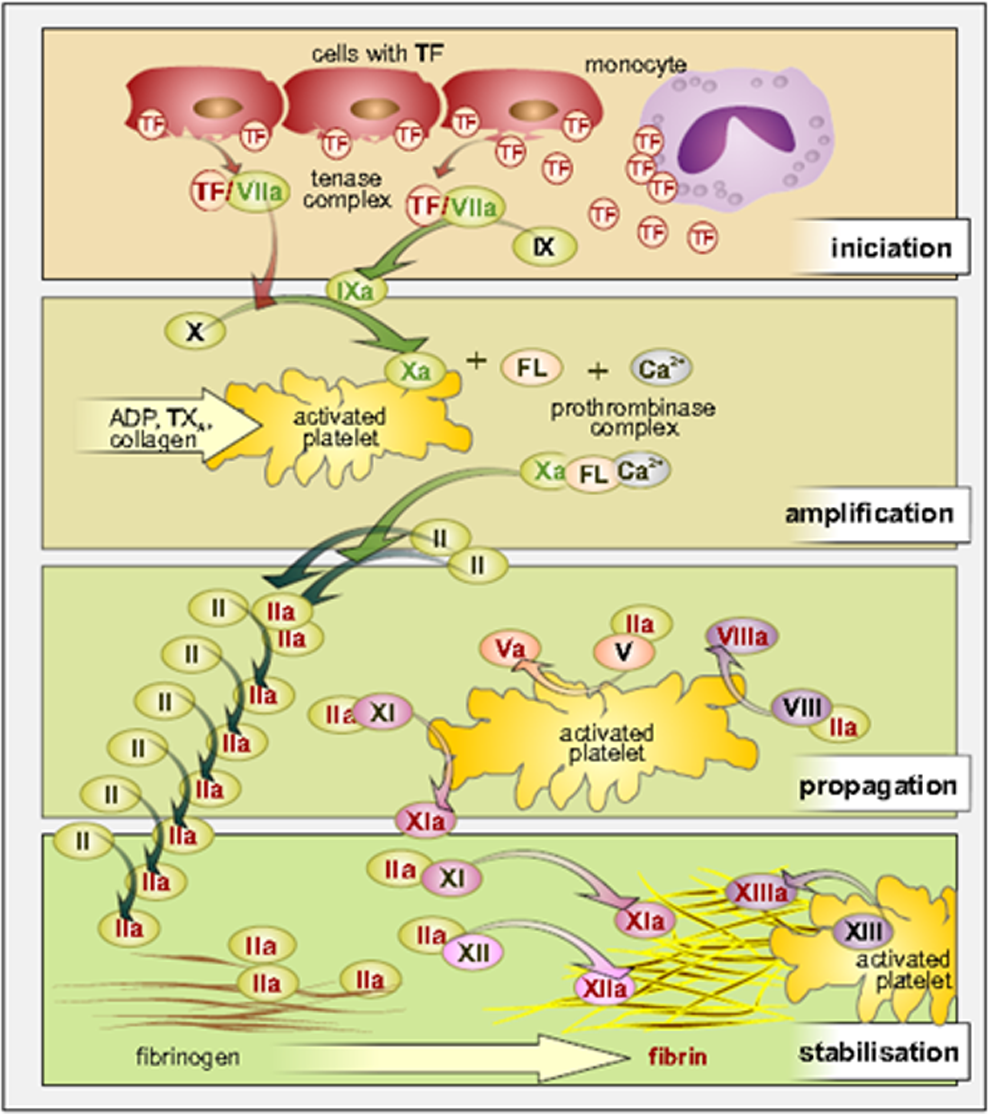
Current view on coagulation cascade. ADP indicates adenosindiphosphate; FL, phospholipids; TF, tissue factor; TX, thromboxane.
In the long term, it is increasingly clear that both extrinsic and intrinsic pathways of the coagulation system are initially triggered by the release of active TF, formation of a complex of TF with FVIIa, and either subsequent direct activation of FX to Xa or via FIXa pathway. The role of FXII in the initiation of the so-called intrinsic coagulation system is currently questioned. Its role has been recently demonstrated mainly in the thrombus propagation together with FXI and in the final stabilization of thrombus together with FXIII. In the search of new antithrombotic agents, the blockade of FXI and FXII could meet the requirement of high efficiency with minimal bleeding complications. 1
The Central Role of TF
The TF (thromboplastin as sometimes incorrectly known—this is a combination of TF and phospholipids; synonym coagulation FIII) has 3 domains—an extracellular, which binds by protein–protein interactions to the FVIIa; transmembranous part; and intracellular portion that mediates signaling functions of the TF. The TF is a transmembrane cellular receptor for FVII/FVIIa, 2 and FVII/ FVIIa is the only ligand that binds to TF and activates both coagulation and signaling pathways.
The TF undergoes posttranslational suppression of procoagulant activity–termed TF encryption, and therefore, most of the TFs expressed on cell surfaces are maintained in a cryptic, that is, inactive state and an activation step (decryption) is required for the expression of maximum TF procoagulant activity. There is no clear explanation of the transition of inactive to procoagulant TF. 3 Decryptic TF binds FVIIa with high affinity, but how the encryption and decryption affect the signaling properties of TF is still unclear. What exactly constitutes cryptic or procoagulant TF, molecular differences between these 2 forms and mechanisms that are responsible for transformation from one to the other form are not entirely clear and remain highly controversial. 4
Except forms bound to the surface of the cell membranes, TF was also found in the circulating microparticles and as an alternatively spliced TF, circulating freely in soluble form, which arises from alternatively spliced TF messenger RNA transcripts lacking exon 5, and hence, exon 4 directly connected to exon 6. Alternatively, spliced human TF circulates in soluble form in blood, contains most of the extracellular domain, and retains the procoagulant properties. 5 Alternatively, spliced human TF exhibits procoagulant activity after contact with the phospholipids and is incorporated into the thrombi and also may contribute to their propagation and stabilization. 5
The TF is expressed on cells in the brain, gut, skin, lung, placenta, and also in the connective tissue cells surrounding blood vessels. The TF thus provides a protective and hemostatic barrier against the external environment if the integrity of the vessels is interrupted. 3 The TF is released also from the ruptured plaque or damaged tissue and may be expressed by monocytes, endothelial cells, macrophages, fibroblasts in response to certain pathological stimuli. The TF expression on the surface of circulating blood cells is very low, but this production can be sharply increased under certain well-defined pathological conditions, such as acute coronary syndrome, inflammation, and cancer propagation. 3,6
Elevated levels of TF were found in patients with inflammation, tumors, or acute coronary syndrome. 7 –11 The TF is also the main activator of the coagulation cascade during viral infection. 12 Congenital deficiency of TF is not known.
The TF, in addition to the initiation of coagulation, participates in signaling in many important biological processes and is considered an important article linking inflammation and thrombosis. The downstream signaling effects of the TF are transduced by activation of protease-activated receptor (PAR)-1 and PAR-2, as well as the PAR-independent pathways, including transactivation of tyrosine kinase receptors. Triggering of signaling cascades such as the mitogen-activated protein kinase and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) signaling pathway couples TF to basic cell functions, such as cell proliferation, migration, and survival. 3
Also, other proteases of the coagulation system participate in cellular responses via PARs. 3 Active TF circulates also in microparticles. Elevated levels of microparticles were found in atherosclerosis, thrombosis, inflammation, metabolic disorders, and tumors. 13,14 Geddings and others have found that TF-positive microparticles enhance thrombosis in mouse models. 15
The primary physiological inhibitor of TF and regulator of coagulation pathway of the FVIIa/TF complex are TF pathway inhibitor (Figure 2). Recently, it was documented that also antithrombin (AT) may act as an important physiological regulator of FVIIa. In vitro studies have shown that AT inhibits FVIIa effectively only if the FVIIa is binding to TF. In this regard, FVII binding to endothelial protein C receptor (EPCR) could play a possible role. 16
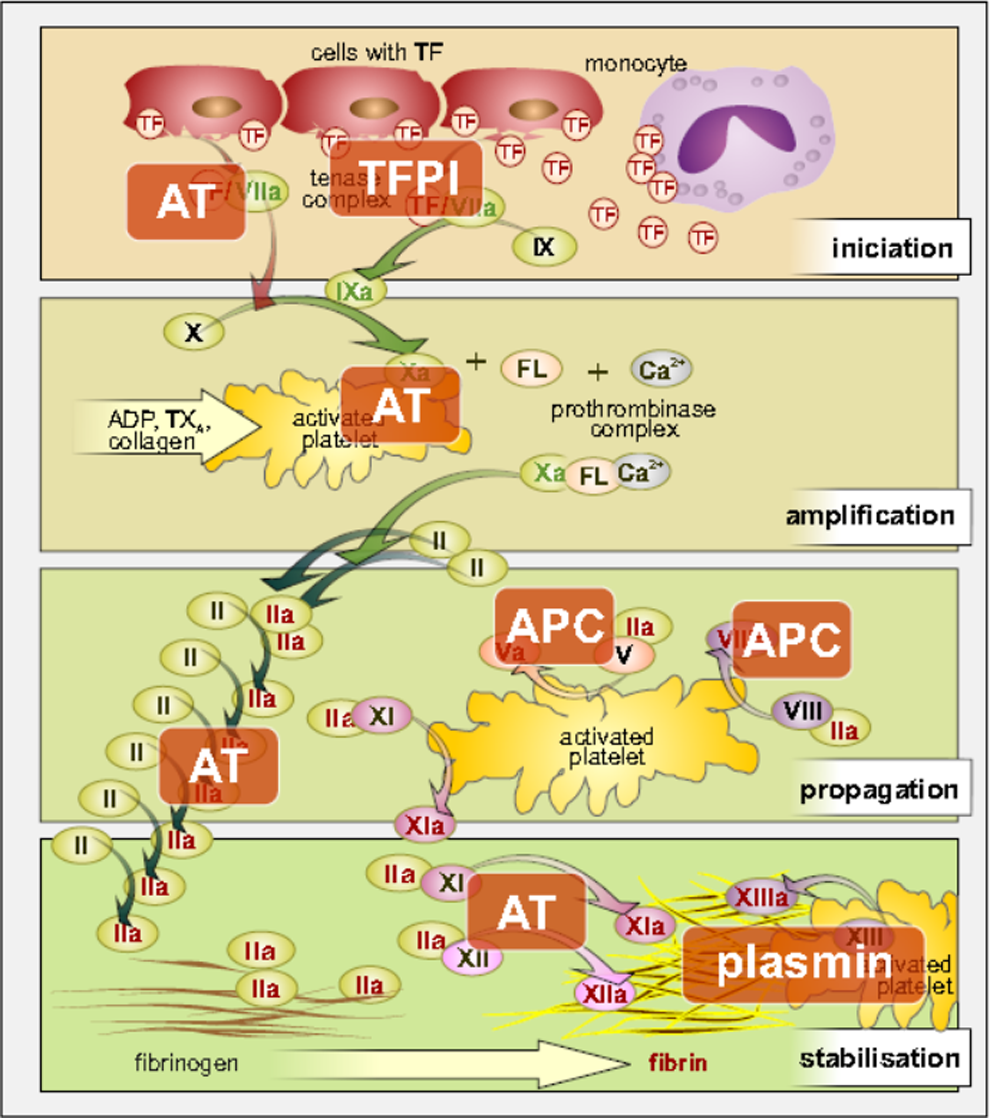
The role of physiological inhibitors of coagulation cascade. ADP indicates adenosindiphosphate; APC, activated protein C—the primary physiological inhibitor; AT, antithrombin; FL, phospholipids; TF, tissue factor; TFPI, tissue factor pathway inhibitor; TX, thromboxane.
The Contact System
The so-called contact system activates laboratory coagulation in vitro; in a humane environment rather serves in the final stages of thrombus formation to its propagation and stabilization. So far, there has been lacking evidence about the role of FXII in physiological hemostasis. The activation of FXII and FXI does not lead to thrombosis and only FXI, but not FXII, deficiency results in slight increase in bleeding tendency with injury (posttraumatic hemorrhage on mucosal surfaces with high endogenous fibrinolytic activity, ie, hemophilia C). 15 Deficiency of FXII is not associated with any bleeding risk. It was shown that neither FXIIa nor FXIa are involved in the in vivo coagulation initiation. Experiments performed recently on animals with the blockade of the contact system (FXII and XI) showed that the contact system is involved in propagation and stabilization of the thrombus, and its blockade reduces the final size of the thrombus in mice, rabbits, and baboons. 13,17 Thrombus formation was impaired under coagulation conditions on plaque material ex vivo in FXII-deficient subject but restored after FXII supplementation. 17 Anti-FXI antibodies as well as FXII and FXI antisense oligonucleotides (which reduce the level of these coagulation factors due to the decreased production in the liver tissue) reduce thrombosis in mice, rats, rabbits, and baboons without an increase in bleeding. 15
Elevated levels of FXI, on the other hand, may be associated with a greater risk of venous thromboembolism and ischemic stroke and possibly with myocardial infarction, whereas reduced plasma levels of FXI offered some protection from venous thromboembolism and stroke but not myocardial infarction. 18 The original concept assumed that trigger of the intrinsic pathway of the coagulation system is a self-activation of FXII on polyanionic surface. High-molecular-weight kininogen (HMWK = Fitzgerald factor) is captured on the specific receptors of endothelial cells and activates prekallikrein (Fletcher factor). Factor XII is activated secondarily by formed prekallikrein, and the process is amplified by reciprocal activation of prekallikrein and FXII. 19 Factor XI can be activated by the same mechanism as prekallikrein. Kallikrein simultaneously cleaves the HMWK to produce bradykinin, which stimulates the release of tissue plasminogen activator. Newer views, however, have considered that the delayed activation of FXII is presumably mediated by components of the atherosclerotic plaque as collagen, nucleic acids, and polyphosphates. 1 Geddings and Mackman in their article proposed that the extracellular RNA bound to both FXII and FXI might be the “long sought after natural foreign surface” for the in vivo activation of the intrinsic pathway, and that there is also evidence that DNA-rich neutrophil extracellular traps and a variety of negatively charged polyphosphates, which include extracellular RNA, DNA, and inorganic polyphosphates that are released during cell damage and infection, activate FXII in vivo. 15
Other Procoagulant and Anticoagulant Factors
Factor V was initially studied in patients with its deficiency, and it was found that these patients were unable to bind FXa to platelets. 20 Factor V was purified and characterized biochemically in 1981. 21 Factor V in its active form (in the original nomenclature, FVa was labeled as FVI) is a cofactor for FXa in the prothrombinase complex, necessary for cleavage of prothrombin to thrombin. Factor V is bound to activated platelets and is activated by thrombin, the heavy and light chains being noncovalently linked by calcium. Like FVIII, FV is not enzymatic in nature. Activated FXa thus requires the presence of calcium and activated FVa in the prothrombinase complex to convert prothrombin to thrombin on the cell membrane.
Factor Va is cleaved by activated protein C (APC). Activation of protein C is important for the negative feedback regulation of coagulation because the APC inactivates both FVIIIa and FVa, the 2 major not enzymatic cofactors responsible for amplification of coagulation leading to the formation of thrombin. This manifests important role of the endothelium as EPCR is necessary to promote protein C activation. 22,23
The EPCR was originally identified and cloned as a specific transmembranous glycoprotein (GP) capable of binding protein C and APC. The EPCR binds protein C and potentiates its activation by thrombin/thrombomodulin complex and thus plays an important regulatory role in the activation of a whole anticoagulant branch of protein C. Adequate production of APC depends on the simultaneous presence of thrombin, thrombomodulin, protein C, and protein C receptor on the surface of endothelial cells. Any change in that composition may lead to a change in the production of APC and thus significantly affect the risk of thrombosis. 23 Thrombin in the presence of thrombomodulin limits its actual activation by a feedback mechanism.
In addition, APC also induces cytoprotective anti-inflammatory and antiapoptotic effects via EPCR-mediated activation of PAR-1. The protein C system plays in this way an important clinical role in the response to trauma, sepsis, inflammatory, and autoimmune disorders. 13
Ligands for EPCR are, apart from protein C and APC, also FVII and FVIIa, Mac-1 on leukocytes, specific variant of γδ T-cell receptor, and P falciparum-infected erythrocyte membrane protein 1. 22 The finding that EPCR acts as a receptor for FVIIa indicates that EPCR may affect hemostasis also by another mechanism than through the APC. It was found that FVIIa binding to EPCR decreases its procoagulant activity by reduction in the formation of FXa via the TF/FVIIa complex pathway. 22 –24
Factor VIII (antihemophilic factor) is an essential coagulation factor, nonenzymatic plasmatic protein, its activated form acts as a cofactor for enzymatic FIXa, and together in the presence of calcium and membrane phospholipids, they form tenase complex that converts FX to its activated form FXa. Factor VIIIa increases the activity of the complex by several orders. Efficacy of tenase complex is controlled either by dissociation of the subunit A1 of FVIIIa or by proteolysis induced by APC along with protein S.
In plasma, FVIII is bound to von Willebrand factor. Liver releases FVIII only into the plasma, which contains von Willebrand factor. Factor VIII is released from this binding on contact with negatively charged phospholipid surfaces and that causes the release of binding site, which was hidden in a complex with von Willebrand factor. Free FVIII is activated by thrombin or FXa.
Plasma levels of FVIII depend among others on the blood group ABO system, which affects the value of some used coagulation tests—activated partial thromboplastin time and thrombin generation assay. 25 Another important factor that contributes to the stabilization of the formed thrombus is FXIII. This fibrin stabilizing factor is necessary for the formation of a network of fibrin fibers. It can be activated in several ways, the main being cleavage of the N-terminal activation peptide on A-subunit by thrombin. 26 Wolberg and Aleman in their laboratory demonstrated that the fibrin or fibrinogen-mediated transport of FXIII in thrombi is necessary for the retention of erythrocytes in the thrombus. 27 Factor XIII stabilizes thrombi against spontaneous fibrinolysis by reciprocal linking of the α2-antiplasmin to fibrin. Cellular FXIII is present in large quantities in platelets and becomes effective after exposure on the surface of activated platelets due to stimulation by thrombin or collagen. 28 The uptake of circulating platelets to the thrombus growing needs several platelet receptors, endothelial matrix components, and coagulation factors. Factor XIII, activated by transglutaminase reaction, covalently binds the von Willebrand factor to polymerizing fibrin. Bound von Willebrand factor further recruits and activates platelets by interaction with the platelet GP Ib receptor. 28 Congenital deficiency of FXIII is associated with life-threatening bleeding, and it is possible to replace the human plasma concentrate or use the recombinant form of FXIII. 29 –32
Coagulation Cascade and the Development of New Antithrombotic Agents
We have witnessed research and rapidly expanding introduction of new antithrombotic agents in the recent years. The experience with the new antithrombotic agents confirmed that the clinical benefit in terms of the ratio between the antithrombotic effect and the risk of bleeding complications depends on which step in the coagulation cascade is the target of intervention. In particular, one can hypothesize that with slight decrease of antithrombotic efficacy, the bleeding tendency might significantly decrease with changing the target of blockade downstream the coagulation cascade (Figure 3). Some support for this assumption is provided by observations concerning the role of FXI and FXII in stabilizing thrombi. Their blockade promises a therapeutic potential for reduction of thrombosis and its complications. 17,33 The advantage of blocking these factors could be the fact that they are necessary for thrombus propagation and stabilization but are not indispensable for hemostasis. Inhibition of FXIIa or reduction of FXI has lowered the incidence of thrombosis of ruptured atherosclerotic plaques in an experiment on mice. 1,17,33 The fact that FXII is activated in vivo by negatively charged polyphosphates, such as extracellular RNA, DNA, and inorganic polyphosphates released during cell damage, has led to the development of nucleic acid-binding polymers as a new class of anticoagulant drug. 15 Although theoretically, the blockade of FXI and XIIa should be effective with minimum of bleeding complications, the clinical efficacy and practical consequences are still to be proven. On the opposite side of this scheme lie TF and FVIIa.
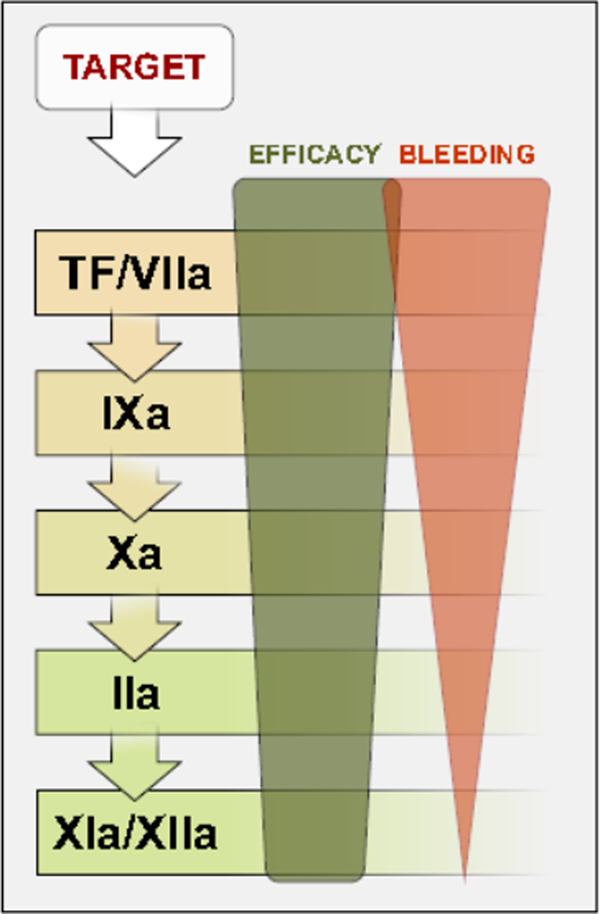
Efficacy and bleeding risk in different categories of pharmacological inhibitors of coagulation cascade. TF indicates tissue factor.
Conclusion
The most significant changes in the contemporary understanding of the coagulation scheme are relating to the unification of the extrinsic and intrinsic pathways under the TF pathway and the new recognition of the role of FXI, FXII, and FXIII in the final stages of thrombus propagation and stabilization.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The preparation of this review was supported by the academic PRVOUK Project P 37/03 Charles University Prague, Czech Republic.