
Abstract
Availability of universal marker for the diagnosis of platelet apoptosis is an important but currently unresolved goal of platelet physiology investigations. Mitochondrial inner transmembrane potential (▵Ψm) depolarization is frequently used as a marker of apoptosis in nucleated cells and anucleate platelets. Since ▵Ψm depolarization in platelets is also frequently associated with concurrent induction of other apoptotic responses, it may appear that ▵Ψm depolarization is a good universal marker of platelet apoptosis. However, data presented in the current study indicate that this is incorrect. We report here fundamental differences in the effects of potassium ionophore valinomycin and calcium ionophore A23187 on human platelet apoptosis. Although both A23187-triggered and valinomycin-triggered ▵Ψm depolarization are strongly induced, the former is dependent on the opening of mitochondrial permeability transition pore (MPTP) and the latter is MPTP-independent. Furthermore, effects of calcium and potassium ionophores on other apoptotic events are also basically different. A23187 induces caspase-3 activation, proapoptotic Bax and Bak protein expression, phosphatidylserine exposure, and microparticle formation, whereas valinomycin does not induce these apoptotic manifestations. Discovery of targeted ▵Ψm depolarization not associated with apoptosis in valinomycin-treated platelets indicates that this marker should not be used as a single universal marker of platelet apoptosis in unknown experimental and clinical settings as it may lead to a false-positive apoptosis diagnosis.
Introduction
The definition of a valid universal marker that may be used for the diagnosis of platelet apoptosis is an important but currently unresolved goal of platelet physiology investigations. A perfect marker should be easily methodically determined and able to predict the presence or absence of platelet apoptosis in unknown tested experimental and clinical conditions.
The role of the mitochondrial pathway in control of apoptosis is well documented for nucleated cells. 1 –3 Dissipation of the mitochondrial inner transmembrane potential (▵Ψm depolarization) determined by cell-penetrating potentiometric dyes is frequently used for detecting apoptosis in nucleated cells 1,2,4 and anucleate platelets. 5
In the review on platelet apoptosis 5 and other papers, 6 –8 many experimental situations are described where ▵Ψm depolarization in platelets has been induced by different triggers, including platelet agonists, calcium (Ca2+) ionophores, proapoptotic BH3 mimetic ABT-737, high shear stresses, and platelet storage. Induction of ▵Ψm depolarization in platelets is associated with concurrent induction of other apoptotic responses, such as opening of mitochondrial permeability transition pore (MPTP), cytochrome c release from mitochondria to the cytosol, activation of caspases-3, -8, and -9, expression of proapoptotic members of Bcl-2 family proteins (Bax, Bak, and Bid), cleavage of apoptosis-associated cytoskeleton proteins (gelsolin, actin, and moesin), phosphatidylserine (PS) exposure on the platelet surface, platelet shrinkage, and platelet-derived microparticle (MP) formation (Table 3 in the review 5 and reseach papers 6 –11 ). Although from these studies 5 –11 it may appear that ▵Ψm depolarization is a good universal marker of platelet apoptosis, data presented in the current report indicate that this is incorrect.
Investigating the effects of potassium (K+) ionophore valinomycin and Ca2+ ionophore A23187 on platelet apoptosis, we demonstrate that, in contrast to Ca2+ ionophore, which induces ▵Ψm depolarization together with other biochemical and cellular manifestations of platelet apoptosis, in platelets treated with K+ ionophore valinomycin, strong ▵Ψm depolarization is not accompanied by the induction of other markers of platelet apoptosis. These data indicate that even strong ▵Ψm depolarization, when detected in unknown tested conditions, cannot be considered an unequivocal marker of platelet apoptosis, and simultaneous determination of other apoptotic markers is required for validation of apoptosis in platelets.
Materials and Methods
Reagents and Solutions
Bovine serum albumin (BSA), dimethyl sulfoxide (DMSO), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), phosphate-buffered saline (PBS), cyclosporin A (CsA), and fluorescein isothiocyanate (FITC)-conjugated F(ab′)2 fragment of goat antirabbit immunoglobulin G (IgG) were purchased from Sigma (St Louis, Missouri). Valinomycin and A23187 were purchased from Calbiochem (San Diego, California). Fluorescent dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide) was purchased from Invitrogen (Carlsbad, California), and FAM-DEVD-FMK (carboxyfluorescein-carbonyl-aspartyl-glutamylvalyl-aspartic acid fluoromethyl ketone) was purchased from Chemicon International (Temecula, California). The FITC-conjugated annexin V was purchased from BD Biosciences (San Jose, California). Rabbit anti-Bax (P-19), anti-Bak (G-23), and anti-Bcl-2 (N-19) IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, California). The FITC-conjugated antiglycoprotein IIb/IIIa (anti-GPIIb/IIIa) antibody (anti-CD41-FITC, clone P2) was purchased from Beckman-Coulter (Westbrook, Maine).
Stock solutions of valinomycin (1.0 mmol/L), A23187 (10 mmol/L), and CsA (10 mmol/L) were dissolved in DMSO. Buffer A was composed of PBS supplemented with 1 mmol/L MgCl2, 5.6 mmol/L glucose, 0.1% BSA, and 10 mmol/L HEPES, pH 7.4. Buffer B was composed of buffer A containing 0.11% DMSO; it was used for dilution of platelet-rich plasma (PRP), preparation of working solutions of valinomycin (5 or 6 μmol/L), A23187 (50 or 60 μmol/L), and CsA (50 μmol/L) for the treatment of platelets, and as the control diluent buffer for the determination of platelet apoptotic events by flow cytometry.
Preparation and Treatment of Platelets with Valinomycin and A23187
Human blood collection and all experimental protocols were approved by the research ethics board of St Michael’s Hospital (Toronto, Ontario, Canada), the methods were carried out in accordance with the approved guidelines, and informed consent was obtained from all participants.
Venous blood from healthy volunteers was anticoagulated with 0.32% sodium citrate. The Platelet-rich plasma (PRP) was obtained by centrifugation at 180g for 15 minutes at room temperature (RT) followed by dilution 1:10 with buffer B (for assays of ▵Ψm, caspase-3, Bax, Bak, and Bcl-2 proteins, and MPs) or 1:60 (for PS assay).
Platelet apoptotic events were determined in 3 platelet groups treated with (1) diluent buffer B, (2) 1 μmol/L valinomycin, 12 and (3) 10 μmol/L A23187. 6,9,11 For this, 40 μL of diluted PRP aliquots was incubated for 15 minutes at RT with 10 μL of buffer B, 5 μmol/L valinomycin in buffer B, and 50 μmol/L A23187 in buffer B, respectively.
Treatment of Platelets with CsA in Combination with Valinomycin, A23187 or Control Diluent
The CsA was used to analyze the effects of MPTP on ▵Ψm depolarization in platelets treated with valinomycin, A23187, or control diluent (buffer B). Six platelet groups were treated at RT with (1) diluent, (2) 10 μmol/L CsA plus diluent, (3) 1 μmol/L valinomycin, (4) 10 μmol/L CsA plus 1 μmol/L valinomycin, (5) 10 μmol/L A23187, and (6) 10 μmol/L CsA plus 10 μmol/L A23187. All platelet treatments were performed with 40 μL of PRP aliquots by dilution of 1:10 with buffer B in a total volume of treatment mixture of 60 μL. For diluent-treated platelet group (1), 40 μL PRP was incubated for 45 minutes with 20 μL buffer B. For CsA plus diluent-treated platelet group (2), 40 μL PRP was preincubated for 30 minutes with 10 μL of 50 μmol/L CsA in buffer B, followed by incubation for 15 minutes with 10 μL buffer B. For valinomycin-treated or A23187-treated platelet groups (3) and (5), 40 μL PRP was preincubated for 30 minutes with 10 μL buffer B, followed by incubation for 15 minutes with 10 μL of 6 μmol/L valinomycin or 60 μmol/L A23187 in buffer B. For CsA plus valinomycin-treated or CsA plus A23187-treated platelet groups (4) and (6), 40 μL PRP was preincubated for 30 minutes with 10 μL of 50 μmol/L CsA in buffer B, followed by incubation for 15 minutes with 10 μL of 6 μmol/L valinomycin or 60 μmol/L A23187 in buffer B.
Determination and Analysis of Platelet Apoptotic Markers
After preparation of appropriately treated platelet samples, platelet apoptosis was determined by flow cytometry (FACSCalibur; BD Biosciences). For this, platelets were incubated with fluorescent probes specific for each of the 7 apoptotic markers, including ▵Ψm depolarization, caspase-3 activation, Bax, Bak, and Bcl-2 protein expression, PS exposure, and MP formation. From each platelet sample, diluted to a final volume of 500 μL with buffer B, 20 000 events were acquired and platelets were detected in the platelet-specific forward scatter (FSC)–side scatter (SSC) gate followed by fluorescence dot plot or histogram analysis using CellQuest Pro, FlowJo 7.4, and FCS Express 3.0 Software (BD Biosciences). Determination of ▵Ψm depolarization in platelets was performed by fluorescent cell-penetrating cationic dye JC-1.
1,9,13,14
The JC-1 working solution was prepared as described previously.
9
Aliquots of platelet samples were incubated with JC-1 solution, acquired for flow cytometry, and platelet-bound JC-1 monomers, detected by green fluorescence 1 (FL1), and JC-1 aggregates, detected by red fluorescence 2 (FL2), were analyzed as FL1-FL2 dot plots. Depolarization of ▵Ψm was determined as the percentage depolarized cells, which is expressed as the increase in the percentage platelets with low FL2.
9
Determination of caspase-3 activation in platelets was performed by flow cytometry using cell-penetrating fluorescein-labeled tetrapeptide FAM-DEVD-FMK as previously described.
9
The FL1 histograms were analyzed and caspase-3 activation was quantified as the mean channel fluorescence (MCF) of platelet-bound FAM-DEVD-FMK probe. Determination of Bax, Bak, and Bcl-2 proteins. Expression of Bcl-2 family proteins was determined by flow cytometry in fixed, permeabilized platelets, using rabbit antihuman anti-Bax, anti-Bak, and anti-Bcl-2 IgG followed by incubation with FITC-conjugated F(ab′)2 fragment of goat antirabbit IgG as previously described.
15
Platelet samples were acquired and gated, and FL1 histograms were analyzed. Bax, Bak, and Bcl-2 proteins were quantified as the MCF of platelet-bound fluorescent F(ab′)2 IgG fragment. Determination of PS exposure on the platelet surface was performed by flow cytometry using FITC-conjugated annexin V. After treatment of platelets with valinomycin, A23187, or diluent buffer B, 45 μL aliquots of platelet samples was mixed with 5 μL of buffer B containing 20 mmol/L CaCl2 to obtain the final CaCl2 concentration of 2 mmol/L and incubated in the dark for 15 minutes at RT with 3 μL annexin V–FITC. Buffer B (450 μL) containing 2 mmol/L of CaCl2 was then added, and samples were acquired, gated, and analyzed by FSC-FL1 dot plots. The PS exposure was determined as the percentage annexin V–positive cells.
9
Determination of platelet-derived MP formation. Enumeration of MPs was performed as described previously in the MP-specific gate using staining of platelet samples with the FITC-conjugated anti-GPIIb/IIIa antibody.
7
Statistical Analysis
Data were analyzed using GraphPad Prism 5 software (GraphPad Software, San Diego, California) and presented as means with standard error of the mean. The statistical significance of the differences between platelet groups was determined by 1-way analysis of variance with Dunnett multiple comparison test and Student t test as appropriate. Differences were considered significant when P < .05.
Results
Mitochondria of nucleated cells respond promptly to the Ca2+ overload, stimulated by Ca2+ ionophore A23187, with a rapid accumulation of the cation in the mitochondrial matrix. 4,16 –18 Calcium influx to mitochondria causes ▵Ψm depolarization, opening of the MPTP in the mitochondrial inner membrane (MIM), followed by mitochondrial matrix swelling, rupture of the MIM and the mitochondrial outer membrane (MOM), and release of proapoptotic proteins from mitochondria to the cytosol. 2,4,17 –20
Treatment of nucleated cells with the K+ ionophore valinomycin facilitates the movement of K+ ions through the MIM down to electrochemical potential gradient, causing ▵Ψm depolarization. 14,21 Valinomycin is highly selective for K+ over sodium (Na+) and Ca2+ ions 22,23 and acts preferentially on the mitochondrial rather than on the plasma membrane. 24 In contrast to mitochondrial Ca2+ influx, stimulated by A23187, mitochondrial K+ influx, stimulated by valinomycin, 24 –27 does not result in MPTP opening in the MIM 19,28 and massive mitochondrial matrix swelling. 19,29
Due to these basic differences in the effects of Ca2+ ionophore A23187 and K+ ionophore valinomycin on mitochondria in nucleated cells, in the current study, we investigated the effects of A23187 and valinomycin on ▵Ψm depolarization and other manifestations of apoptosis in anucleate platelets.
Effects of Valinomycin and A23187 on Platelet ▵Ψm Depolarization
As shown in Figure 1, both A23187 and valinomycin strongly induce ▵Ψm depolarization in platelets, when 57.8% ± 8.4% and 94.0% ± 1.1% of cells, respectively, have the MIM with dissipated membrane potential, in comparison with 12.5% ± 4.0% of cells in the control diluent-treated platelets (P < .001).
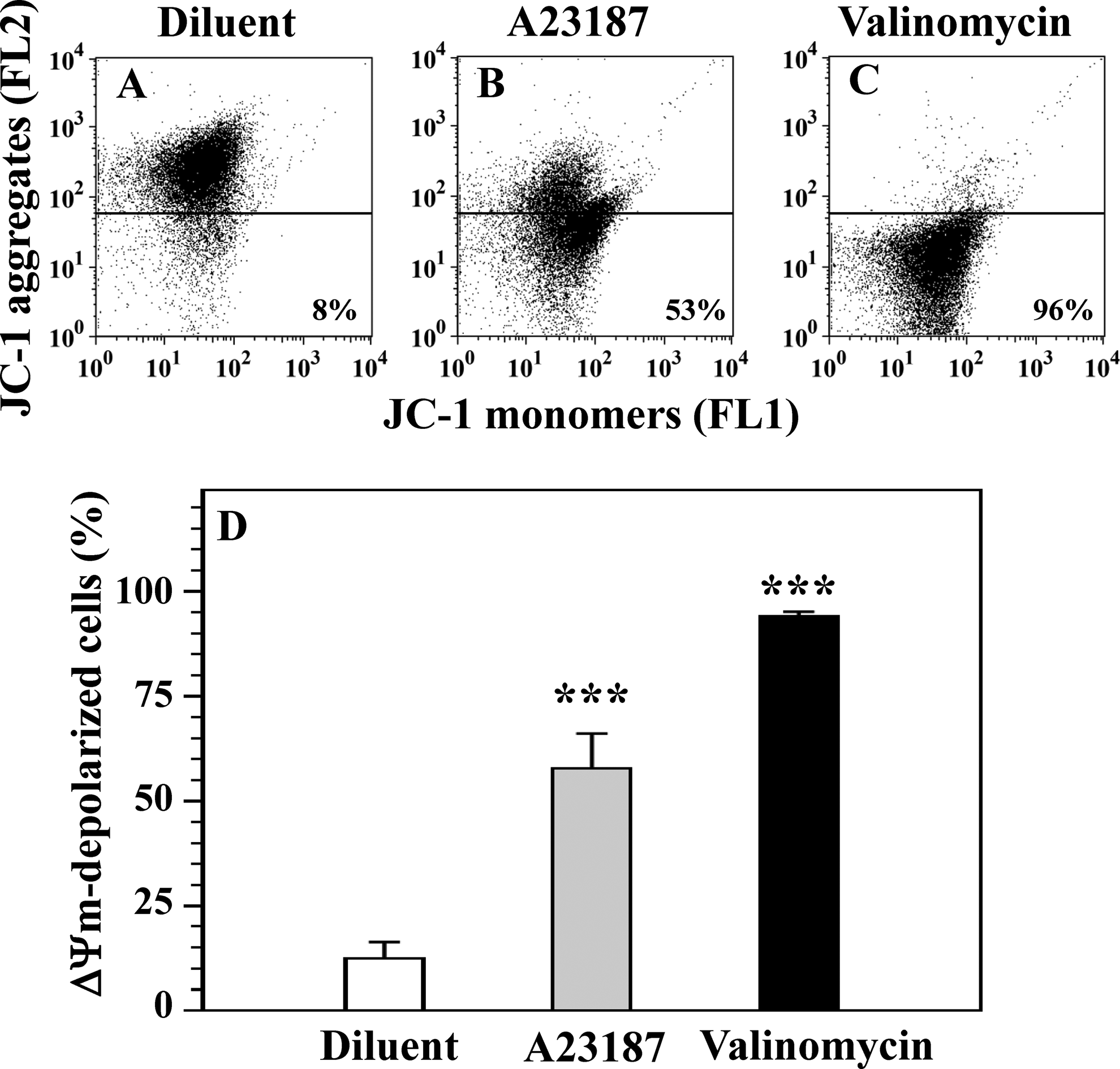
Calcium ionophore A23187 and potassium ionophore valinomycin induce dissipation of the mitochondrial inner membrane potential (▵Ψm depolarization) in human platelets. Platelets were treated with diluent buffer B (A), 10 μmol/L A23187 (B), and 1 μmol/L valinomycin (C), and ▵Ψm depolarization was determined by fluorescent cell-penetrating cationic dye JC-1. Note that ▵Ψm depolarization in A23187-treated and valinomycin-treated platelets is characterized by increase in the percentage cells with low platelet-bound red fluorescence (FL2) of JC-1 aggregates (B and C), in comparison with that in diluent-treated platelets (A). Numbers below horizontal lines in panels (A-C) represent the percentage depolarized cells. Means and SEM for 6 to 7 experiments, and differences between A23187-treated and valinomycin-treated platelet groups versus diluent-treated platelet group are presented: ***P < .001(D). SEM indicates standard error of the mean.
Dependence of Valinomycin-Induced and A23187-Induced ▵Ψm Depolarization on the MPTP Opening in Platelets
Using CsA, a potent and specific MPTP inhibitor, 1,2,8,9,30,31 we investigated the role of the MPTP opening in platelet ▵Ψm depolarization triggered by Ca2+ and K+ ionophores. As shown in Figure 2, A23187-induced ▵Ψm depolarization is completely inhibited by pretreatment of platelets with CsA, indicating that depolarization induced by Ca2+ ionophore is dependent on MPTP opening. In contrast, we found that ▵Ψm depolarization induced by the K+ ionophore valinomycin is not inhibited by CsA and independent on MPTP opening (Figure 2).
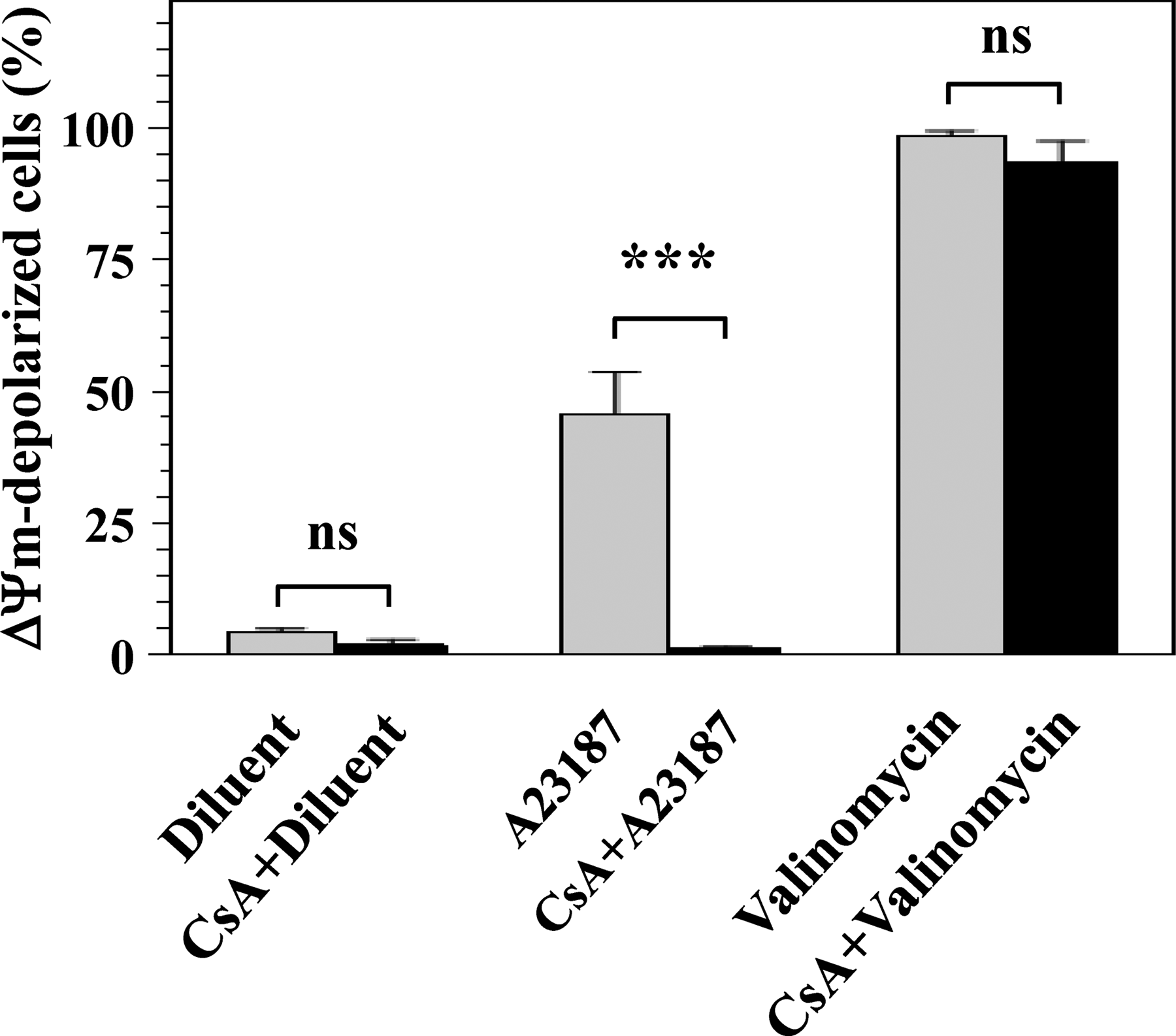
The MPTP-dependent and MPTP-independent ▵Ψm depolarization in platelets treated with calcium ionophore A23187 and potassium ionophore valinomycin. The role of MPTP in platelet ▵Ψm depolarization triggered by A23187 and valinomycin was studied using CsA, a potent inhibitor of MPTP opening. For this, platelets were treated with diluent buffer B, 10 μmol/L A23187, and 1 μmol/L valinomycin or pretreated with 10 μmol/L CsA before treatments with diluent, A23187, and valinomycin. The ▵Ψm depolarization was determined by JC-1 assay as described in Figure 1. Means and SEM and differences between CsA-treated versus CsA-untreated platelet groups are presented for 3 experiments. Note that CsA significantly inhibits A23187-induced ▵Ψm depolarization (***P < .001), indicating the dependence of A23187-triggered depolarization on MPTP opening. In contrast, valinomycin-induced ▵Ψm depolarization is not inhibited by CsA (ns: P > .05), indicating that depolarization triggered by valinomycin is independent on MPTP opening. CsA indicates cyclosporin A; MPTP, mitochondrial permeability transition pore; ns, nonsignificant; SEM, standard error of the mean; ▵Ψm, transmembrane potential of the mitochondrial inner membrane.
Effects of Valinomycin and A23187 on Extra-Mitochondrial Manifestations of Platelet Apoptosis
We further demonstrated that the effects of A23187 and valinomycin on extra-mitochondrial biochemical and cellular manifestations of platelet apoptosis are also fundamentally different (Figures 3 –6). Treatment of platelets with Ca2+ ionophore induces activation of apoptosis executioner caspase-3 (Figure 3), expression of proapoptotic Bax and Bak proteins, but not antiapoptotic Bcl-2 protein (Figure 4), and PS exposure on the platelet surface (Figure 5). On the contrary, K+ ionophore does not induce these biochemical apoptotic events (Figures 3 –5). Furthermore, A23187 strongly induces MP formation, a cellular manifestation of platelet apoptosis, whereas valinomycin does not stimulate MP formation (Figure 6).
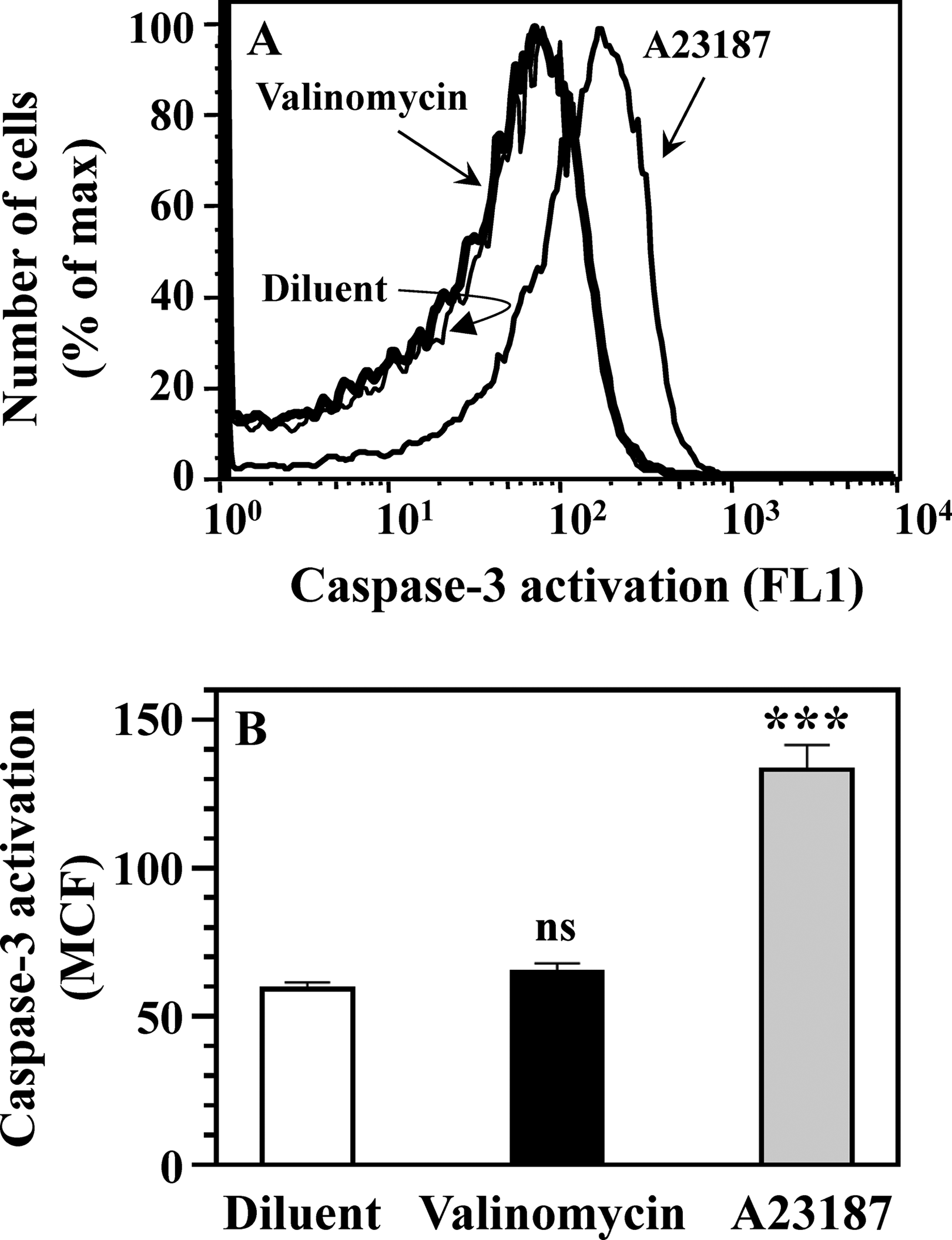
Caspase-3 activation in platelets treated with potassium ionophore valinomycin and calcium ionophore A23187. Platelets were treated with diluent buffer B, 1 μmol/L valinomycin, and 10 μmol/L A23187, and caspase-3 activation was determined by fluorescent cell-penetrating FAM-DEVD-FMK probe, specific for active caspase-3. Binding of FAM-DEVD-FMK to active caspase-3 was analyzed by FL1 histograms (A) and quantified by determining the MCF of caspase-bound FAM-DEVD-FMK (B). Means and SEM for 10 experiments and differences between A23187-treated and valinomycin-treated platelet groups versus diluent-treated platelet group are presented (B). Note that A23187 significantly induces caspase-3 activation (***P < .001), whereas valinomycin does not induce activation of caspase-3 (ns: P > .05). This result indicates that even strong ▵Ψm depolarization (Figure 1) does not promote activation of apoptosis executioner caspase-3 in valinomycin-treated platelets. FL1 indicates fluorescence 1; MCF, mean channel fluorescence; SEM, standard error of the mean; ▵Ψm, transmembrane potential of the mitochondrial inner membrane.
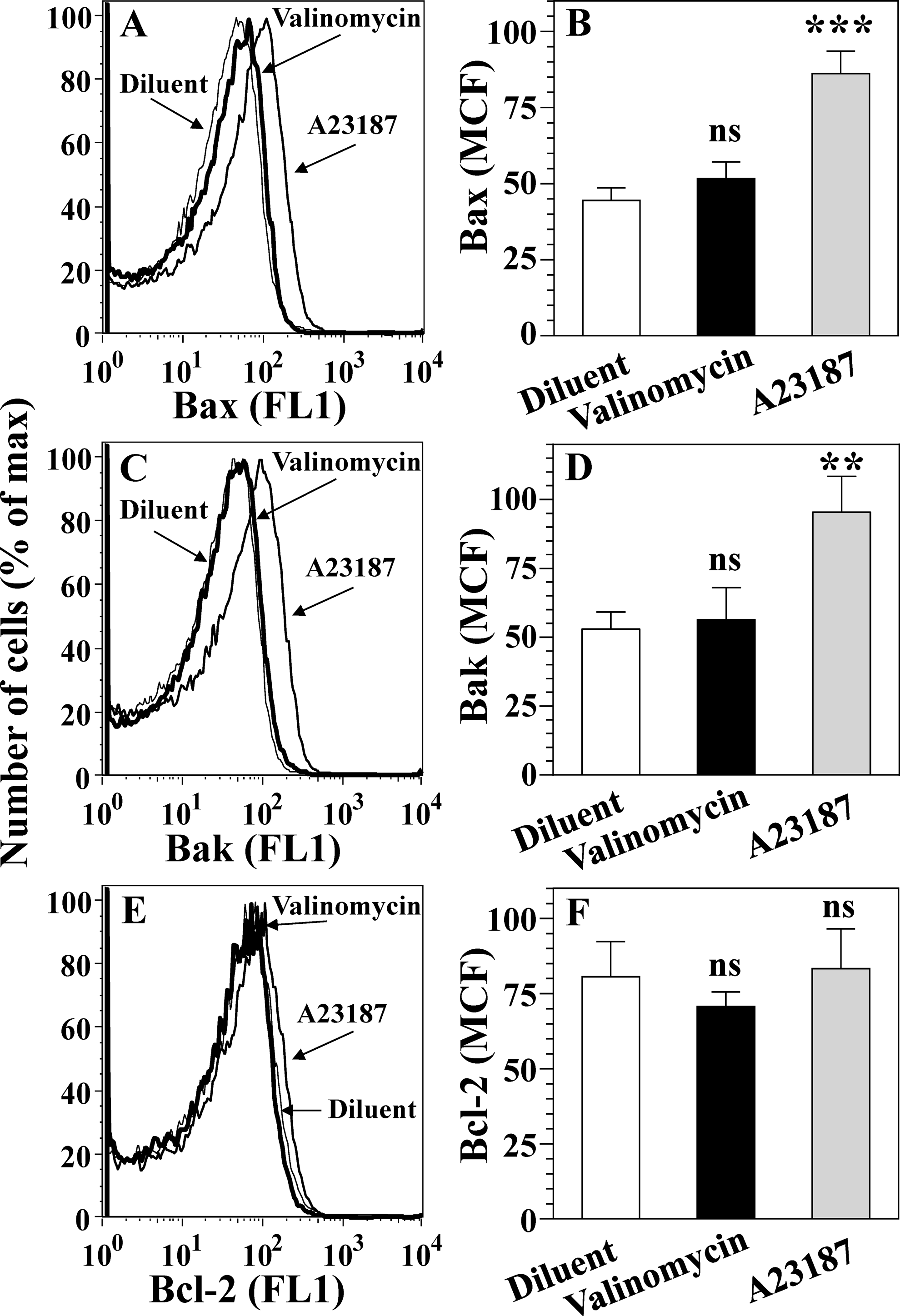
Expression of proapoptotic Bax and Bak proteins and antiapoptotic Bcl-2 protein in platelets treated with potassium ionophore valinomycin and calcium ionophore A23187. Platelets were treated with diluent buffer B, 1 μmol/L valinomycin, and 10 μmol/L A23187, fixed and permeabilized. The Bcl-2 family proteins were determined using anti-Bax, anti-Bak, and anti-Bcl-2 IgG followed by anti-IgG fluorescent probe. Expression of Bax, Bak, and Bcl-2 proteins was analyzed by FL1 histograms (A, C, E) and quantified by determining the MCF (B, D, F). Means and SEM for 6 to 7 experiments and differences between A23187-treated and valinomycin-treated platelet groups versus diluent-treated platelet group are presented (B, D, F). Note that A23187 significantly induces expression of proapoptotic Bax (***P < .001) and Bak (**P < .01) proteins, whereas valinomycin does not induce expression of these proteins (ns: P > .05; B and D). Both A23187 and valinomycin do not affect the expression of antiapoptotic Bcl-2 protein (E and F). This result indicates that strong ▵Ψm depolarization (Figure 1) does not promote the expression of proapoptotic Bax and Bak proteins in valinomycin-treated platelets. FL1, indicates fluorescence 1; IgG, immunoglobulin G; MCF, mean channel fluorescence; ns, nonsignificant; SEM, standard error of the mean; ▵Ψm, transmembrane potential of the mitochondrial inner membrane.
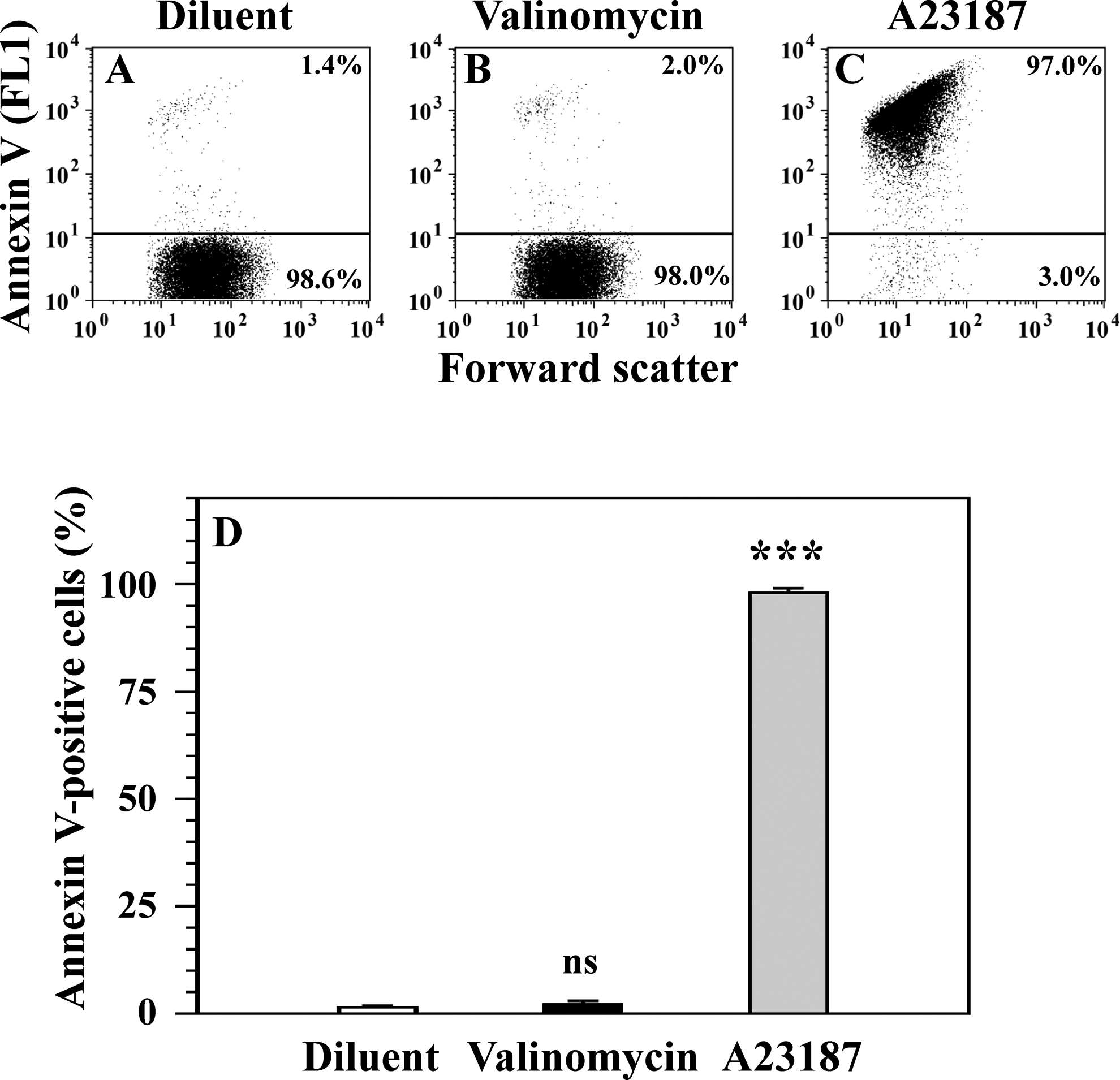
Exposure of PS on the surface of platelets treated with potassium ionophore valinomycin and calcium ionophore A23187. Platelets were treated with diluent buffer B (A), 1 μmol/L valinomycin (B), and 10 μmol/L A23187 (C), and PS exposure was determined by FITC-conjugated annexin V (FL1). Numbers above horizontal lines in panels (A-C) represent the percentage cells with exposed PS. Means and SEM for 10 experiments and differences between A23187-treated and valinomycin-treated platelet groups versus diluent-treated platelet group are presented (D). Note that A23187 significantly and strongly induces aberrant PS exposure on the platelet surface (***P < .001), whereas valinomycin does not induce PS exposure (ns: P > .05). This result indicates that strong ▵Ψm depolarization in valinomycin-treated platelets (Figure 1) does not promote PS exposure. FITC indicates fluorescein isothiocyanate; FL1, fluorescence 1; PS, phosphatidylserine; SEM, standard error of the mean; ▵Ψm, transmembrane potential of the mitochondrial inner membrane.
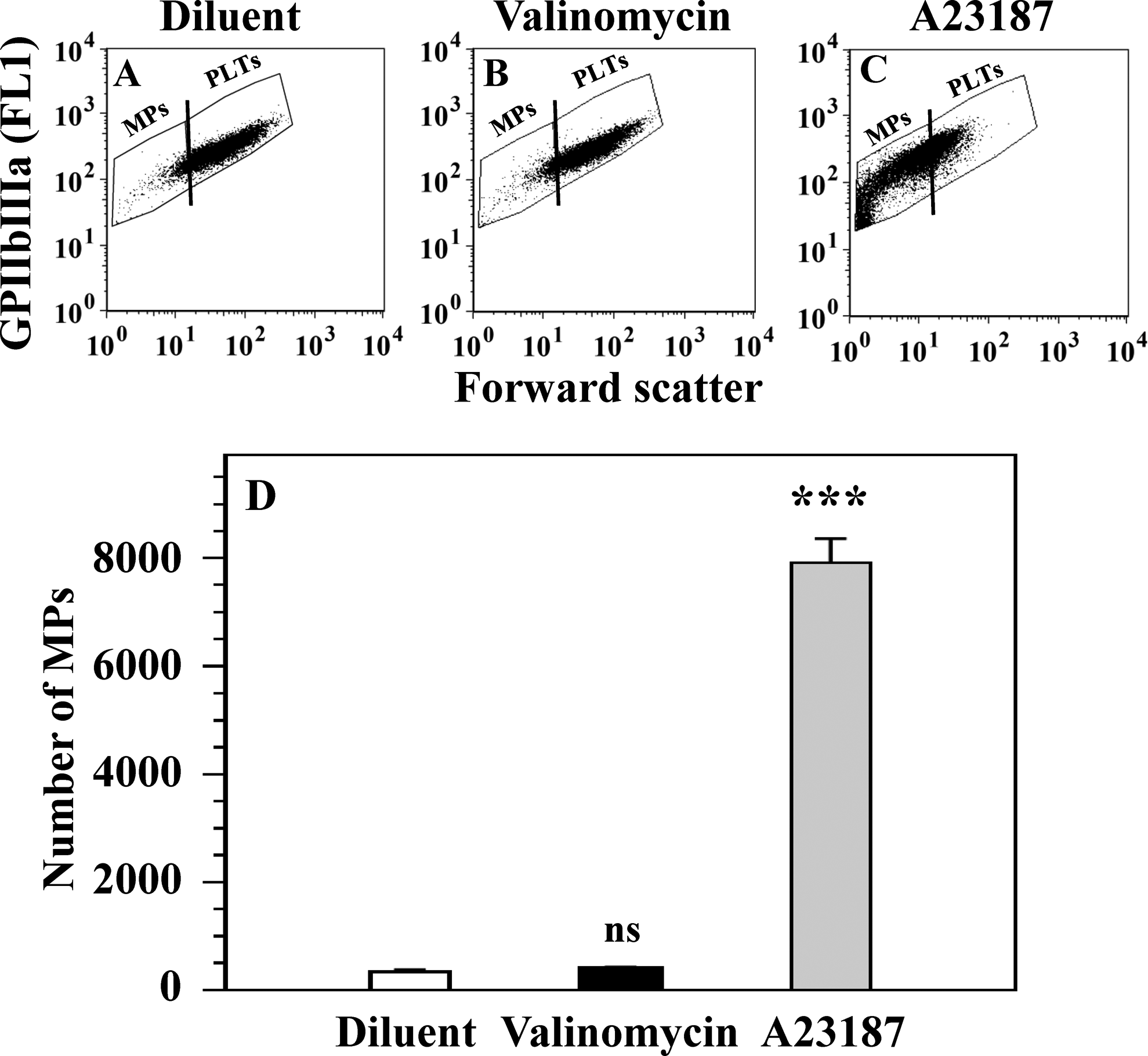
Formation of MPs in platelets treated with potassium ionophore valinomycin and calcium ionophore A23187. Platelets were treated with diluent buffer B (A), 1 μmol/L valinomycin (B), and 10 μmol/L A23187 (C). Platelet-derived MPs were determined in the MP-specific gate in platelet samples stained with FITC-conjugated anti-GPIIb/IIIa antibody. The MPs were numerated on the left of vertical lines in forward scatter-GPIIb/IIIa (FL1) dot plots (A-C). Means and SEM for 4 experiments and differences between A23187-treated and valinomycin-treated platelet groups versus diluent-treated platelet group are presented (D). Note that A23187 strongly induces the formation of MPs (***P < .001), whereas valinomycin does not induce the MP formation (ns: P > .05). This result indicates that strong ▵Ψm depolarization in valinomycin-treated platelets (Figure 1) does not promote MP formation. FITC indicates fluorescein isothiocyanate; FL1, fluorescence 1; MP, microparticle; SEM, standard error of the mean; ▵Ψm, transmembrane potential of the mitochondrial inner membrane.
Discussion
Effects of Valinomycin and A23187 on ▵Ψm Depolarization, MPTP Opening, and Other Manifestations of Apoptosis in Platelets and Nucleated Cells
Table 1 summarizes the results of the current study on the effects of Ca2+ ionophore A23187 and K+ ionophore valinomycin on mitochondrial (Figures 1 and 2) and extra-mitochondrial biochemical (Figures 3 –5) and cellular (Figure 6) manifestations of platelet apoptosis. As shown in the Table 1, both Ca2+ and K+ ionophores strongly induce ▵Ψm depolarization in human platelets. However, the mechanisms involved in the stimulation of ▵Ψm depolarization by these ionophores are different. The Ca2+ ionophore induces ▵Ψm depolarization by MPTP-dependent process through MPTP opening in the MIM. In contrast, ▵Ψm depolarization induced by K+ ionophore is independent of MPTP opening (Figure 2). Furthermore, A23187 induces extra-mitochondrial biochemical and cellular manifestations of platelet apoptosis, including caspase-3 activation, expression of proapoptotic Bax and Bak proteins, PS exposure, and MP formation, whereas valinomycin does not induce these apoptotic events. Hence, we demonstrate for the first time that the K+ ionophore valinomycin accomplishes only targeted triggering of ▵Ψm depolarization but does not induce platelet apoptosis.
Different Impact of Calcium Ionophore A23187 and Potassium Ionophore Valinomycin on Mitochondrial and Extra-Mitochondrial Manifestations of Platelet Apoptosis.
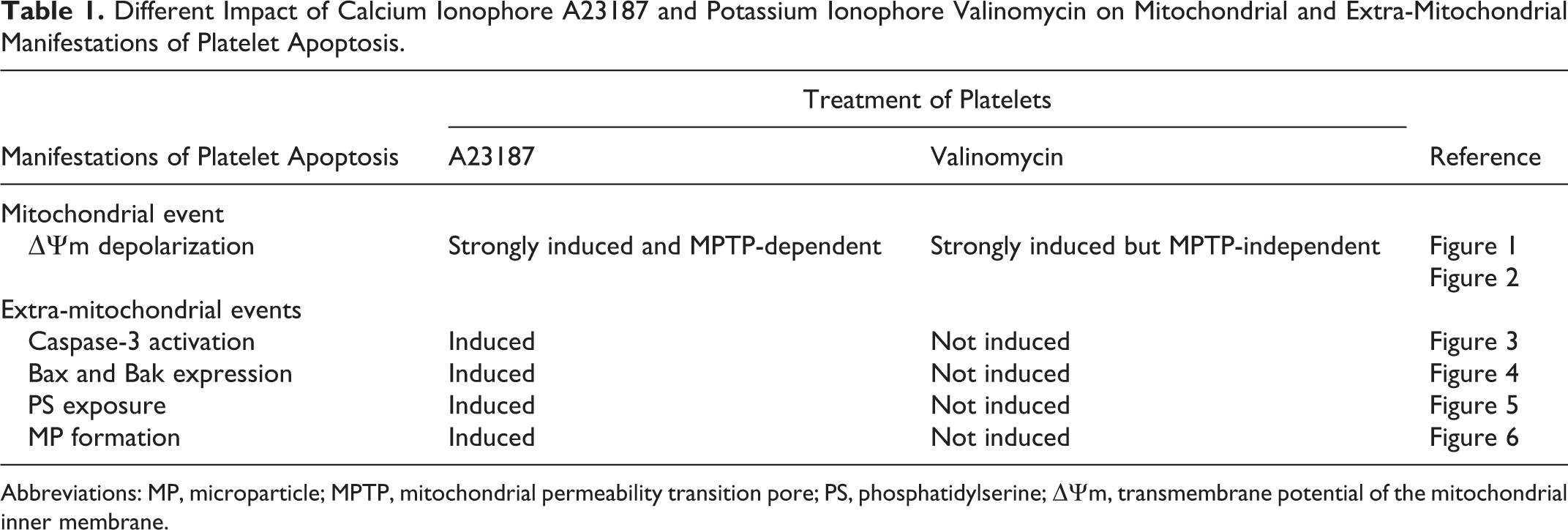
Abbreviations: MP, microparticle; MPTP, mitochondrial permeability transition pore; PS, phosphatidylserine; ▵Ψm, transmembrane potential of the mitochondrial inner membrane.
Figure 7 presents the results obtained in this study together with the data reported by others on the effects of these ionophores on the processes associated with the intrinsic (mitochondrial) pathway of apoptosis in nucleated cells, including (1) mitochondrial Ca2+ and K+ influxes, (2) MPTP opening in the MIM, (3) mitochondrial matrix swelling, (4) MIM and MOM rupture, and (5) release of proapoptotic proteins from mitochondrial matrix and/or mitochondrial intermembrane space (IMS) to the cytosol (references a-j in Figure 7 legend). Figure 7A shows that the mitochondrial Ca2+ overloading influx induced by A23187 4,16 –18 triggers strong MPTP-dependent ▵Ψm depolarization in platelets, which is inhibited by CsA and mediated by MPTP opening in the MIM. By contrast, as seen in Figure 7B, the strong ▵Ψm depolarization triggered by valinomycin-induced K+ influx 24 –27 is MPTP-independent, not inhibited by CsA, and occurs in the absence of MPTP opening. As shown in Figure 7A, data obtained on isolated mitochondria of nucleated cells treated with Ca2+ demonstrated MPTP opening in the MIM, 2,4,17,18,20 massive mitochondrial matrix swelling, MIM and MOM rupture, 2,4,17,19,20 and release of proapoptotic proteins both from IMS and mitochondrial matrix. 4,17,19 In contrast, valinomycin-treated nucleated cell mitochondria (Figure 7B) are characterized by the absence of MPTP opening, 19,28 no massive matrix swelling and the absence of MIM and MOM rupture, 19,29 and no or limited release of proapoptotic proteins from IMS but not from the mitochondrial matrix. 19
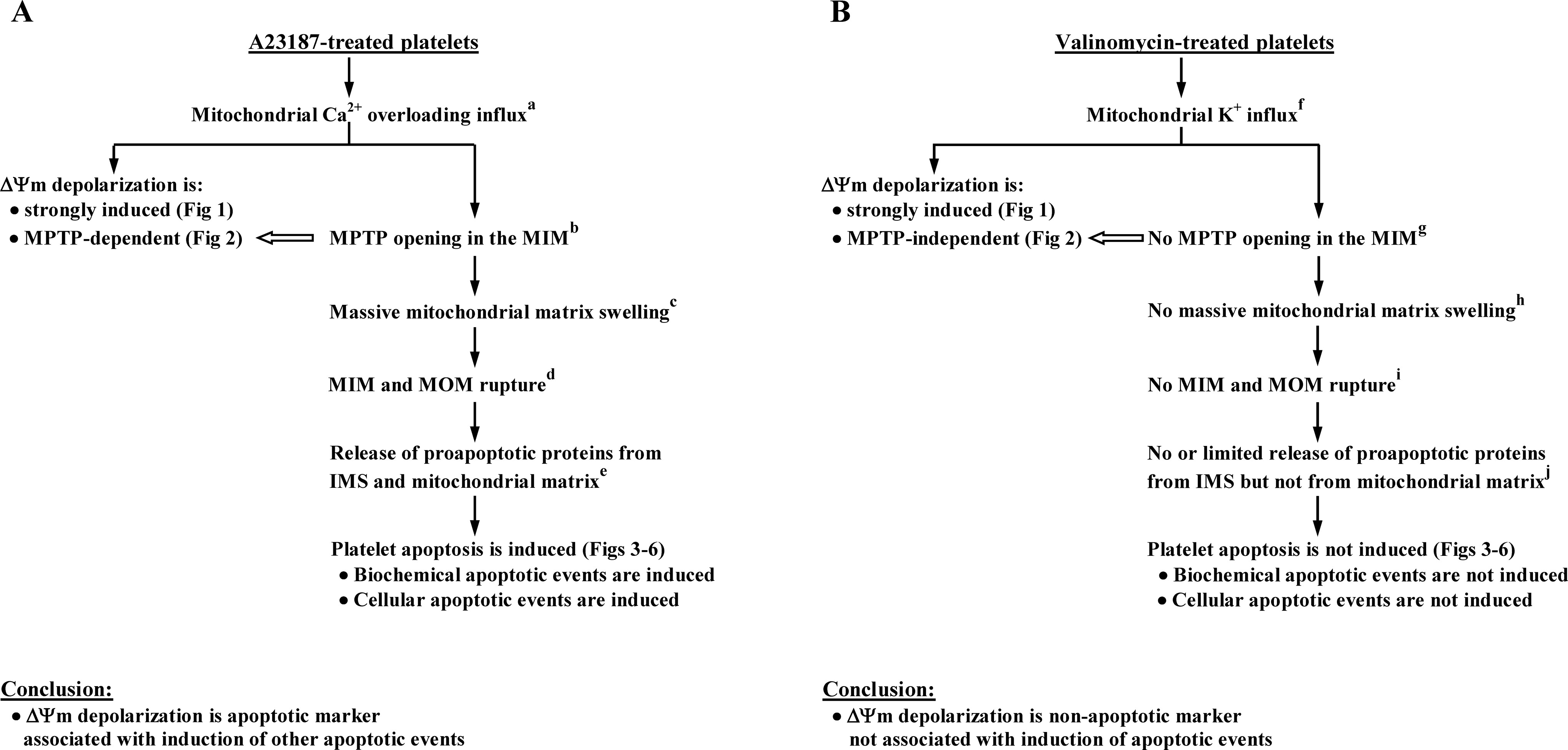
The ▵Ψm depolarization: apoptotic (A) and nonapoptotic (B) platelet marker. The results obtained in the current study for A23187-treated (A) and valinomycin-treated (B) platelets are presented (Figures 1 –6), as well as data obtained by others in nucleated cells (referencesa-j). Note that effects of mitochondrial calcium (Ca2+) influx induced by Ca2+ ionophore A23187 and potassium (K+) influx induced by K+ ionophore valinomycin on ▵Ψm depolarization and other manifestations of platelet apoptosis are fundamentally different. (A and B), Both Ca2+ and K+ influxes trigger strong ▵Ψm depolarization in platelets (Figure 1), but A23187-induced ▵Ψm depolarization is MPTP-dependent (Figure 2), whereas valinomycin-induced ▵Ψm depolarization is MPTP-independent (Figure 2). Mitochondrial Ca2+ influx induces biochemical and cellular manifestations of platelet apoptosis (A: Figures 3 –6), whereas K+ influx does not induce platelet apoptosis (B: Figures 3 –6). Experiments performed in nucleated cells demonstrate that mitochondrial Ca2+ influxa induces MPTP opening in the MIM,b massive mitochondrial matrix swelling,c MIM and MOM rupture,d and release of proapoptotic proteins from IMS and mitochondrial matrixe (A), and K+ influxf, in contrast, does not induce these eventsg-j (B). Taken together, these data indicate that, depending on triggering stimulus, ▵Ψm depolarization in platelets may be apoptotic (A) or nonapoptotic marker (B) associated or not associated with other manifestations of apoptosis. References in (A): a 4,16 –18 , b 2,4,17,18,20 , c,d 2,4,17,19,20 , e 4,17,19 . References in (B): f 24 –27 , g 19,28 , h,i 19,29 , j 19 . IMS indicates intermembrane space between MIM and MOM; MIM, mitochondrial inner membrane; MOM, mitochondrial outer membrane; MPTP, mitochondrial permeability transition pore; ▵Ψm, transmembrane potential of the MIM.
However, it has been reported that effects of valinomycin and other K+ ionophores on apoptosis in nucleated cells are dependent on cell type. Valinomycin-treated Chinese hamster ovary cells exhibit several apoptotic events, including ▵Ψm depolarization, caspase-3 activation, and PS exposure. 32 In murine hematopoietic cells, valinomycin triggers ▵Ψm depolarization, and inhibition of MPTP prevents subsequent valinomycin-induced ▵Ψm depolarization. 33 In rat ascites hepatoma (AH-130) cells, valinomycin induces caspase-3 activation and cell shrinkage, as well as uncoupling of respiration and ▵Ψm depolarization, suggesting that this type of tumor cells may reflect a mechanism transmitting the signal from ▵Ψm depolarization to subsequent execution steps of apoptosis. 34 In primary mouse microglia and astrocytes and cell lines (BV-2, C6, and HEK 293), on the other hand, valinomycin promotes an alternative autophagic form of cell death rather than apoptosis. 35 In the erythroleukemic K562 cell line, the K+ exchanger nigericin and K+ ionophore valinomycin trigger ▵Ψm depolarization, and the pretreatment of cells with CsA fails to protect K562 cells from the loss of ▵Ψm induced by intracellular K+ flux manipulations. 28
During the treatment of platelets by valinomycin, this K+ ionophore may have other effects beyond ▵Ψm depolarization. It may be speculated that alternative forms of platelet cell death occur in platelets treated with valinomycin. As in nucleated cells, other consequences of ▵Ψm depolarization may occur in anucleate platelets, including energy starvation by uncoupling of respiration and ▵Ψm depolarization 34 and the autophagic form of cell death rather than apoptosis. 35 To examine these options, oxidative phosphorylation and autophagy may be tested in valinomycin-treated platelets downstream of ▵Ψm depolarization.
Conclusions and Biomedical Significance
From the above characterization of the relationships between ▵Ψm depolarization and other mitochondrial (MPTP opening) and extra-mitochondrial manifestations of apoptosis in platelets treated with Ca2+ ionophore A23187 and K+ ionophore valinomycin, we make the following conclusions.
Differing Effects of Mitochondrial Ca2+ and K+ Influxes on Platelet Apoptosis
We show a fundamental difference between the impact of mitochondrial Ca2+ influx, induced by Ca2+ ionophore A23187, and mitochondrial K+ influx, induced by K+ ionophore valinomycin, on platelet apoptosis. Since strong ▵Ψm depolarization can occur both in the presence (A23187 treatment) and in the absence (valinomycin treatment) of MPTP opening (Figure 2), it appears that ▵Ψm depolarization does not require participation of MPTP-dependent mechanism of the mitochondrial membrane permeabilization. Furthermore, it was shown that Ca2+ influx triggers platelet apoptosis (Figure 7A), whereas K+ influx does not cause apoptosis (Figure 7B).
Depolarization of ▵Ψm is not a Universal Marker of Apoptosis in Anucleate Platelets
Presented data demonstrate that strong ▵Ψm depolarization can be achieved by both modes of platelet treatment—with Ca2+ and K+ ionophores. In the case of treatment with Ca2+ ionophore A23187, ▵Ψm depolarization triggered by mitochondrial Ca2+ overloading influx is associated with induction of other manifestations of platelet apoptosis and represents apoptotic marker (Figure 7A). However, with valinomycin-treated platelets, ▵Ψm depolarization is not associated with platelet apoptosis and cannot be considered as an apoptotic marker (Figure 7B). Hence, ▵Ψm depolarization is not a universal marker of platelet apoptosis but, depending on experimental and clinical conditions, may also represent a nonapoptotic marker characterizing electrochemical potential of the MIM not relevant to platelet apoptosis.
Discovery of Nonapoptogenic ▵Ψm Depolarization in Platelets
Our experiments performed on valinomycin-treated platelets demonstrate that ▵Ψm depolarization per se is not an apoptogenic factor (ie, this mitochondrial event is not causal for platelet apoptosis), since even strong valinomycin-induced ▵Ψm depolarization, when almost all cells in platelet population have depolarized MIM (Figure 1), does not trigger biochemical and cellular manifestations of apoptosis in anucleate platelets (Table 1).
Permeabilization of the MIM as an Apoptogenic Factor in Platelets
Mitochondrial inner membrane permeabilization rather than ▵Ψm depolarization may be a primary apoptogenic factor of platelet apoptosis. Previously, we found that pretreatment of A23187-stimulated platelets with MPTP inhibitor CsA completely prevents A23187-induced ▵Ψm depolarization, caspase-3 activation, platelet shrinkage, and MP formation and partially inhibits PS exposure. 9 The results of the current study (Figures 1 –6) and data obtained from mitochondria of nucleated cells (references a-j in Figure 7) together suggest that permeabilization of the MIM executed via MPTP opening (if followed by massive mitochondrial matrix swelling, MIM and MOM rupture, and release of proapoptotic proteins from IMS and mitochondrial matrix to the cytosol), rather than ▵Ψm depolarization, may be a primary apoptogenic event of intrinsic pathway of platelet apoptosis, which determines execution or nonexecution of apoptosis in A23187-treated and valinomycin-treated platelets, respectively (Figure 7).
Diagnosis of Platelet Apoptosis by ▵Ψm Depolarization
The current work indicates that the detection of platelet ▵Ψm depolarization in unknown tested experimental and clinical conditions is insufficient for the diagnosis of platelet apoptosis, and determination of other apoptotic markers is required in order to avoid a false-positive conclusion.
Targeted Triggering of ▵Ψm Depolarization by Valinomycin as a Model for Analyzing Platelet Functions and Clearance
Targeted valinomycin-induced ▵Ψm depolarization in platelets may be a useful experimental model for studying the role of ▵Ψm depolarization in different platelet reactions and functions, including platelet hemostatic function and platelet clearance.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the grants to VL-T6285 from the Heart and Stroke Foundation of Ontario, Canada, and Platelet Research Fund of Ronya Beskin, Israel.