
Abstract
The prevalence of factor V (FV) Leiden G1691A, prothrombin G20210A, and methylenetetrahydrofolate reductase (MTHFR) C677T mutations were investigated among 90 sickle trait, 61 sickle homozygous, 75 sickle beta thalassemia, and 15 HbSD Asian Indian sickle cell patients. In all, 297 healthy controls were evaluated to compare the polymorphism frequency. The prevalence of FV Leiden heterozygous G>A were significant in the group (P = .02), while PRT G20210A polymorphism was not seen among patients as well as controls. However, an increased frequency of the MTHFR 677 C>T genotype was seen among patients as well as controls, but this was not statistically significant (P = .13). This suggested a low impact of inherited hypercoagulability risk factors in the pathogenesis of sickle cell disease and/or its complications.
Introduction
Sickle cell disease (SCD) is one of the most common inherited diseases in the world. In India, 10 000 individuals with SCD are reported per million populations. Even with the same inherited DNA mutation within the β-globin gene, patients present a notorious clinical heterogeneity. 1 Additional genetic factors such as α-thalassemia, fetal hemoglobin synthesis, and β-globin haplotype have been identified, but none of these can fully explain the differences in clinical expression observed in these patients. Probably, there are other elements that contribute to the many phenotypes of the disease. 2–5 It has been known for a long time that patients with SCD show activation of the blood coagulation and fibrinolytic systems, as well as increased platelet activity and consumption of coagulation inhibitors, especially during vaso-occlusive crises and also during the steady state of the disease. 6–9 Vascular complications are an important and perplexing aspect of the clinical spectrum of sickle cell anemia, although there is controversial evidence surrounding the role of thrombosis in this complication. 10,11 Because of the importance of vascular complications in the pathophysiology of SCD, a number of genetic polymorphisms associated with thrombophilia have been studied as potential genetic modifiers of SCD. Inherited risk factors for vascular disease include factor V (FV) Leiden (G1691A), 12 prothrombin (PRT) G20210A, 13 and methylenetetrahydrofolate reductase (MTHFR) C677T point mutations 14 ; and in view of their role in enhancing thrombus formation, it was suggested that these mutations play a role in the pathogenesis of SCD 15,16 and/or its complications. It has been hypothesized that this subset may have some other underlying genetic predisposition. A possible pathogenetic mechanism is thrombophilia. Glueck et al 17 found that 74% of their patients (none of whom had SCD) with osteonecrosis had one or more primary coagulation disorders, the most common of which were resistance to activated protein C (APC) and hypofibrinolysis or a combination of both. Hyperhomocysteinemia, which can result from a variety of enzymatic defects in the biochemical pathway of folate metabolism, is an independent risk factor for premature vascular disease. 18–20 A C>T substitution at nucleotide 677 in the MTHFR gene is associated with a thermolabile phenotype and diminished enzyme activity with consequent hyperhomocysteinemia. Studies among patients with SCD from Brazil 16 and the United States 15,21 have not demonstrated an association with ischemic stroke; however, Kutlar et al 22 identified this MTHFR mutation as a risk factor in the development of avascular necrosis (AVN) among their patients. No authentic data have been reported among Asian Indians. In this study, the prevalence of these 3 point mutations was assessed in patients with SCD and healthy controls from Asian Indian origin.
Materials and Methods
Study participants were 241 sickle cell patients and 297 age- and sex-matched controls and all were Indians. Five milliliters of venous blood were collected after obtaining the signed informed consent form. Study was approved by the institutional ethic committee. All sickle subtype was diagnosed by cation exchange high-performance liquid chromotography, and complete blood count analysis was done by Sysmex auto analyzer.
Detection of Polymorphism
Total genomic DNA was isolated from peripheral blood leukocytes by the kit (bioserve) method. Factor V Leiden, PRTG20210A, and MTHFR C677T genotype analysis was performed by polymerase chain reaction–restriction fragment length polymorphism analysis using MnlI 12, HindIII 13, and HinfI 14 digestion (all procured from New England Biolabs, Beverly, Massachusetts). Statistical analysis was performed using GraphPad statistics software. Yates chi-square test was used to assess the intergroup significance.
Result
The study participants were divided into 5 groups; group 1 comprised 90 sickle trait (52 male and 38 female with a mean age of 22.27
Among the sickle traits, 3 were heterozygous (G>A) and none was a homozygous (A>A) carrier of FV Leiden; while in sickle homozygous patients, 4 were heterozygous and 1 was homozygous. Among patients with sickle beta thalassemia, 8 were heterozygous and 1 was homozygous; while in sickle D patients, 2 were heterozygous and 1 was homozygous. Higher frequency of FV Leiden gene was found in sickle D patients (20%) while among controls, 73 were heterozygous and 27 were homozygous carrier.
Eleven heterozygous of MTHFR C677T found in sickle trait, while 13 heterozygous and 4 homozygous in HbSS (Sickle cell anemia) patients. In patients with sickle beta thalassemia, 16 were heterozygous and 4 homozygous; while, in contrast, in patients with sickle D, 2 were heterozygous, a significant increase in MTHFR C677T carrier rate (27.6%) was seen in sickle homozygous patients compared to healthy participants where 37 were heterozygous and 3 were homozygous (13.5%). No frequency of heterozygous PRT G20210A was found in any of the patient groups and controls. However, the frequency of the FV Leiden (G1691A) and MTHFR (C677T) point mutations was higher among patients when compared to controls, but this did not reach statistical significance. All the details are given in Table 1 .
Frequency of thrombotic mutations in sickle cell patients
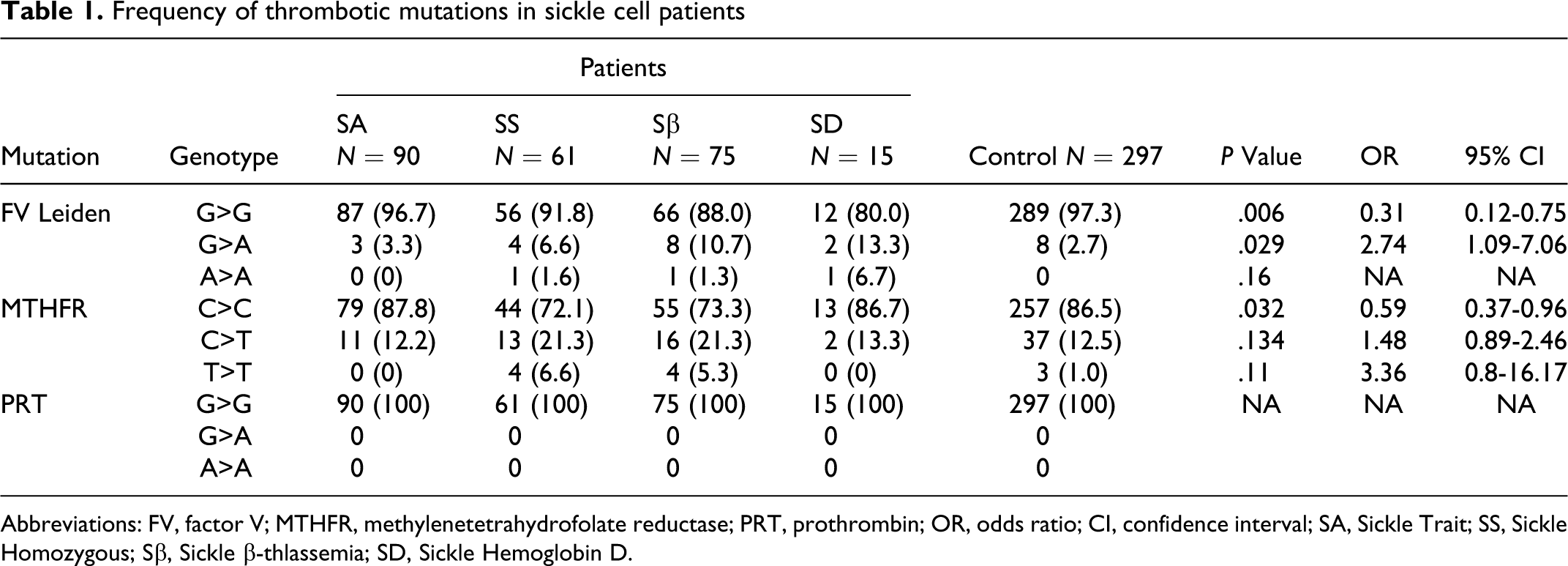
Abbreviations: FV, factor V; MTHFR, methylenetetrahydrofolate reductase; PRT, prothrombin; OR, odds ratio; CI, confidence interval; SA, Sickle Trait; SS, Sickle Homozygous; Sβ, Sickle β-thlassemia; SD, Sickle Hemoglobin D.
Discussion
Several genetic factors contribute to the phenotypic diversity of SCD, and genetic polymorphisms associated with thrombophilia were implicated as genetic modifiers of SCD. 15 The importance of thrombosis in the pathophysiology of SCD has been recently recognized. Insofar as FV Leiden, PRT G20210A, and MTHFR C677T were implicated in the pathogenesis of thrombotic events, we assessed their prevalence among patients with SCD. Low frequencies of FV Leiden (1.90%) and PRT G20210A (0.0%) were seen among healthy individuals in Eastern Saudi Arabia (Dammam), 23 in agreement with a study on their prevalence in Western Saudi Arabia (Jeddah), where rates of 2.2% and 1.1% were reported for FV Leiden and PRT G20210A, respectively. 24 Considerable evidence suggests that endothelial damage and thrombosis play important contributory roles in the vaso-occlusive complications of SCD. 10,25 Among all the groups, 3 patients with HbS were homozygous FV Leiden and 17 patients with sickle cell were heterozygous FV Leiden, and 8 in control group were heterozygous FV Leiden . Heterozygous FV Leiden frequency was statistically significant (P = .02). Prothrombin (PRT G20210A) polymorphism was not detected in any group of patients and controls. MTHFR C677T appears to play a role in SCD, evidenced by increased prevalence of T>T (and C>T) genotypes among patients with SCD. The MTHFR 677TT genotype was detected in 3.31% of HbS patient group while 1.22% among controls; 17.42% of heterozygous MTHFR 677C>T (17.42%) was detected in sickle patient group and 15.35% in the controls. Although MTHFR 677T gene frequency is high in patients and controls, this did not reach statistical significant level. Other investigator reported the prevalence 15,26–28 in the range of 1% to 2.8%. Balasa et al 27 found a prevalence of 1% for this genotype among 110 normal American Blacks, whereas its prevalence among Caucasians ranges from 10% to 20%, depending on the geographic distribution. 29–34 In Brazil, despite the great miscegenation, there seems to be a high proportion of Caucasians within normal individuals, and Morelli et al 35 reported a 9% prevalence of 677TT and Franco et al 36 found a 16.6% prevalence. Some investigators did not find an association between MTHFR C677T and clinical manifestations in SCD. According to the previous reports, FV Leiden and PRT gene mutations were found at a high frequency in some European regions and were very rare in African descendents. In Brazil, reports have described a low frequency in the general population, varying from 1% to 2% and 0.7% to 3.6% for the G1691A and G20210A mutations, respectively. The frequency of the 677TT allele (MTHFR) in Brazil is high among Caucasians (10.3%) but low in African descendents (1.4%) and Indians (1.2%). On the other hand, reports from the State of São Paulo presented very different results for the frequency of the 677TT allele in patients with SCD, with none in Campinas and 1.8% in São Paulo. 16,37,38 In French Canadians, 19 it has been reported as 38%while in a Brazilian study, it was found to be 36% in whites, 40% in Asians, 24% in Amerindians, 5% in African Blacks, and 12% in Brazilian Blacks. 39 Two studies among African American patients with SCD reported frequencies of 9% and 15%, respectively. 16,22
Conclusion
In conclusion, our data suggest a low impact of inherited hypercoagulability risk factors in the pathogenesis of SCD and/or its complications. Sickle cell disease is generally mild among patients probably because the patients carry the Saudi Arabia/India bS-globin gene cluster haplotype that is associated with a high Hb F level. Both thrombosis and the sickle cell anemia have contradict pathophysiology that makes a mild phenotype of the disease.
Footnotes
Acknowledgments
The authors thank the technical staff of the Department of Hematology, AIIMS, for expert assistance.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: supported by ICMR & Hematology Department AIIMS, New Delhi, India.