
Abstract
Keywords
Introduction
There are several low-molecular-weight heparins (LMWHs) available with the brand name including dalteparin (Pfizer, US), enoxaparin (Sanofi-Aventis, France), nadroparin (GlaxoSmithkline, UK), certoparin (Novartis, Switzerland), reviparin (Abbott, US), and parnaparin (Alpha-Wasserman, Italy) with variable availability in different countries. Each of these LMWHs has a characteristic molecular weight profile and biological activity in terms of an anti-factor Xa and anti-factor IIa potency, and clinical effect. It is now widely accepted that individual LMWHs are chemically unique agents and cannot be interchanged therapeutically. 1 Each commercial LMWH has been individually developed for specific clinical indications, which are dose and product dependent. Recently, several generic LMWHs have become available worldwide, and companies have filed for regulatory approval of generic versions of enoxaparin, without head-to-head clinical trials, based on compound biochemical characteristics, particularly Anti-factor Xa activity in healthy volunteers. The Food and Drugs Administration (FDA) has suggested that immunogenicity should also be considered as an important evaluation for biological compounds. 2 The FDA has also recently approved a generic version of enoxaparin for the US market, pending approval for another one. 3 As the primary aim of a generic drug is to reduce costs without compromising patient care, a generic drug is required to be chemically and biologically equivalent to the pioneer drug. Because LMWHs represent biological drugs, clinical trials should be used to determine generic equivalency to the branded drug. The available literature supports the fact that there is no correlation between Anti-factor Xa, anti-factor IIa activity, and clinical outcome. 4 Multiple generic versions of enoxaparin were previously tested in animal models to determine safety, efficacy, and pharmacodynamic parameters. 5 In order to prove that generic LMWHs are clinically equivalent to branded LMWHs in humans, an exploratory randomized clinical trial comparing these 2 therapies as prophylaxis for venous thromboembolic events following major abdominal surgery was conducted.
Methods
Patient Population
The study was conducted in 4 different sites in Brazil (Appendix A). Inclusion criteria were age >18 years and patients undergoing major abdominal surgery with formal indication of chemoprophylaxis for venous thromboembolism (VTE) according to the seventh American College of Chest Physicians (ACCP) guidelines. 6
Exclusion criteria were pregnancy, active clinically significant bleeding, recent gastrointestinal bleeding or documented congenital bleeding disorder/disorders, thrombocytopenia (platelet count below 100 × 109/L), hepatic or renal dysfunction, uncontrolled hypertension (blood pressure 200 mm Hg systolic and/or 110 mm Hg diastolic), acute bacterial endocarditis, a history of hemorrhagic stroke, recent (<2 months prior to randomization) brain, spinal, or ophthalmologic surgery, or a known hypersensitivity to heparin or LMWH. Also excluded were patients that participated in any other therapeutic study evaluating deep vein thrombosis (DVT) prophylaxis trial in the last 90 days. The Institutional Review Boards of the 4 sites involved in this research approved the study protocol. All patients signed a written informed consent before entry to the study, which was conducted according to the guidelines for good clinical practice (GCP). 7
Study Design
A total of 200 consecutive patients were randomized in a 1:1 ratio to received either 40 mg of Sanofi-Aventis branded enoxaparin (SAe) subcutaneously ([sc] 4000 IU) or 40 mg of eurofarma-enoxaparin ([Ee] sc; 4000 IU) started either preoperatively or till 6 hours after surgical procedure according to the physician discretion and then given once daily (od) postoperatively from 7 to 10 days. Randomization performed on a 1:1 basis, with the first patient receiving one of the study drugs, randomly assigned, and then alternating drugs with each subsequent patient. Enrollment initiated in January 2005 with last patient last visit (LP/LV) occurred in August 2007.
Primary end points were safety (major bleeding) and efficacy (composite of symptomatic DVT, proximal asymptomatic DVT, pulmonary embolism [PE], and VTE-related death) clinically relevant nonmajor bleeding (CRNM) and minor bleeding. Bilateral compressive ultrasound (CUS) of the legs, including venous iliac system was performed in all patients on day 10 + 4. (For CUS technique and bleeding definitions, see Appendix A.)
Follow-ups including clinical and biochemical evaluations were performed till 60 days after surgery. In addition, clinical findings occurring at any time that were suggestive of a venous thromboembolic event were evaluated with CUS, and contrast computed tomography (CT), and/or pulmonary scintigraphy if necessary.
Further end points included the occurrence of thrombocytopenia, defined as a platelet count below 100 000/mm3 and/or a minimum decrease of 40% compared to baseline.
Anti-factor Xa activity was performed in the initial 20 patients, during the trial. Due to concerns regarding heparin contamination by oversulfated chondroitin sulfate (OSCS), nuclear magnetic resonance (NMR) for the detection of OSCS was also performed on the study drugs (Appendix A). This information did not impact the outcome of the study since in 2005-2007, the problem of heparin contamination was not known publicly.
Sample Size Calculation/Statistical Analysis
Sample size was calculated based on an estimated occurrence of primary end points in a range of 2.5% to 10% (bleeding/safety) and in 5% to 15% (VTE events/efficacy) of patients. With a confidence interval (CI) of 95%, an α = 5% and power of 70%, 180 patients were enough to demonstrate safety of the study drug in this exploratory trial. We elected 200 patients to enroll in this study.
Statistical analysis was performed on an intention-to-treat (ITT) basis, thus including all patients who received at least one dose of the study drug. Univariate analysis (Student t test) and chi-square tests were used for the statistical analysis of demographic and clinical parameters. Comparisons were performed within the 2 enoxaparin groups regarding demographics, clinical and biochemical examinations, primary, and secondary end points using Bonferroni multiple comparisons. Unilateral tests were used for the safety analysis and bilateral tests for efficacy end points. Microsoft Excel was used for data management and table analysis. A confidence interval of 95% was set with a P value of less than .05 considered statistically significant. For anti-factor Xa comparative activity, an all pairwise multiple comparison procedures (Student-Newman-Keuls Method) and analysis of variance (ANOVA) were performed.
The Consolidated Standards of Reporting Trials (CONSORT) diagram of the study enrollment and follow-up is shown in Figure 1 .
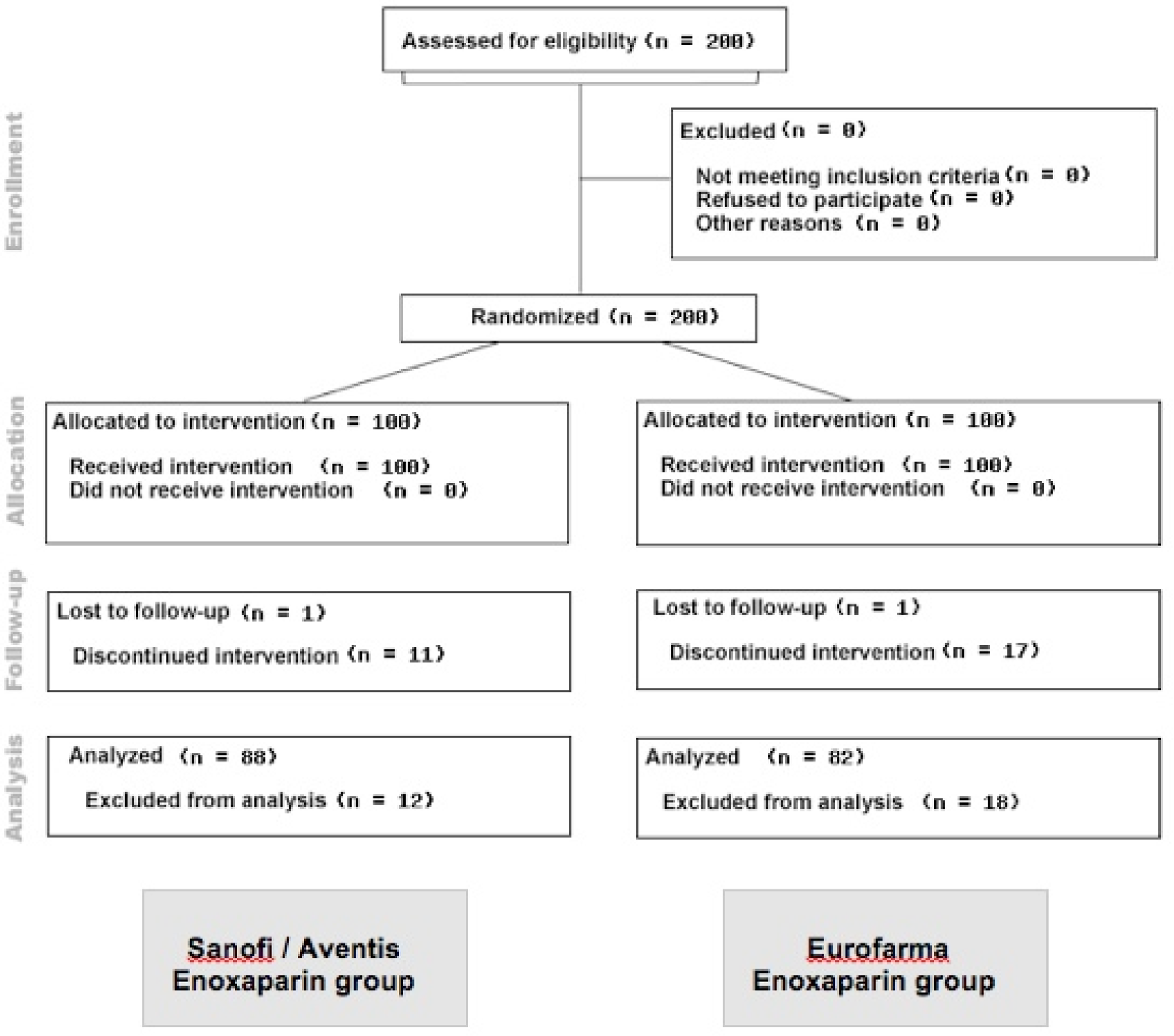
Flow chart of Consolidated Standards of Reporting Trials (CONSORT).
Results
The groups were very homogeneous with regard to gender, age, race, weight, and height (Table 1 ).
Baseline Characteristics a
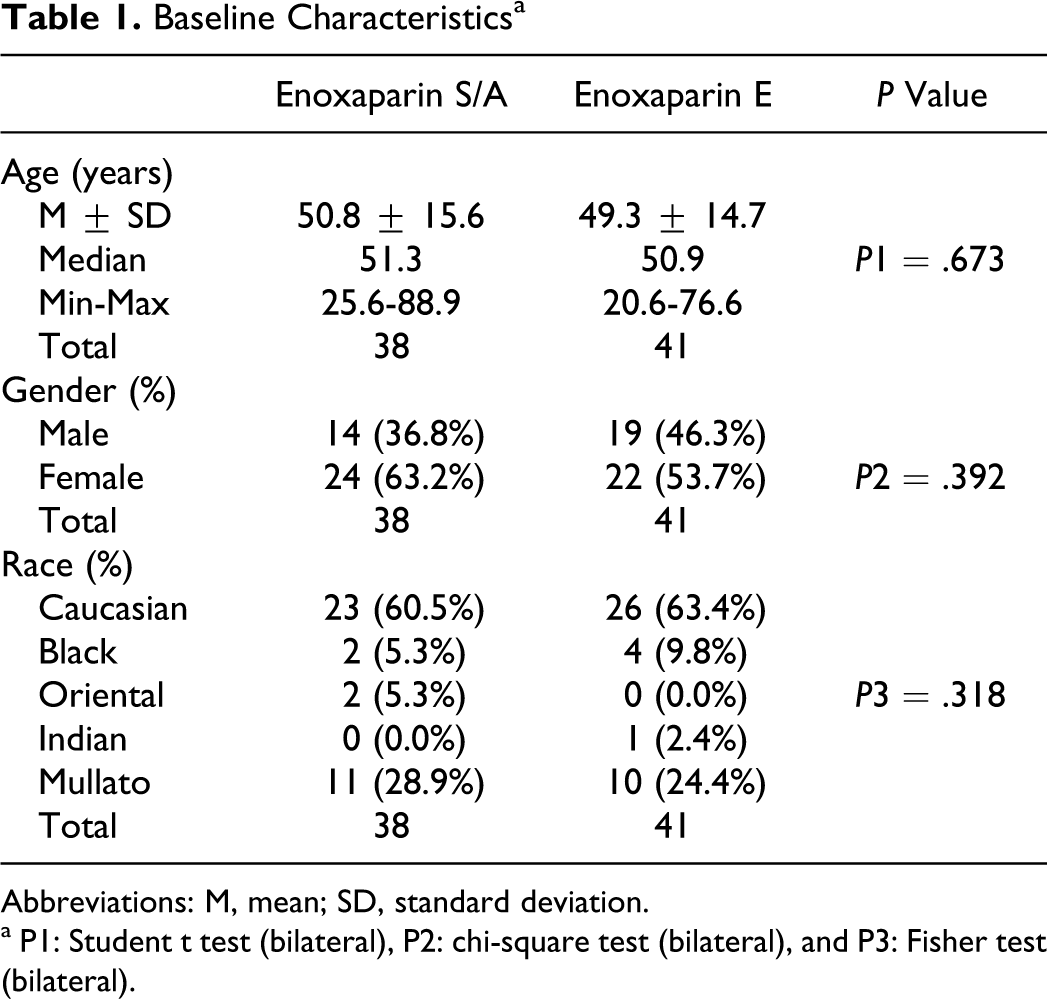
Abbreviations: M, mean; SD, standard deviation.
a P1: Student t test (bilateral), P2: chi-square test (bilateral), and P3: Fisher test (bilateral).
Eighty-five percent of the patients enrolled were followed up according to the protocol (15% were not captured due to withdrawal of informed consent or lack of follow-up). No statistically significant differences between the 2 groups were detected in any of the measured outcomes: no major bleeding events occurred in either group (Table 2 ). Two patients in the SAe group developed DVT, while none of the Ee group experienced DVT (Table 3 ).
Adverse Events Including Bleeding (Primary Safety End Point)a
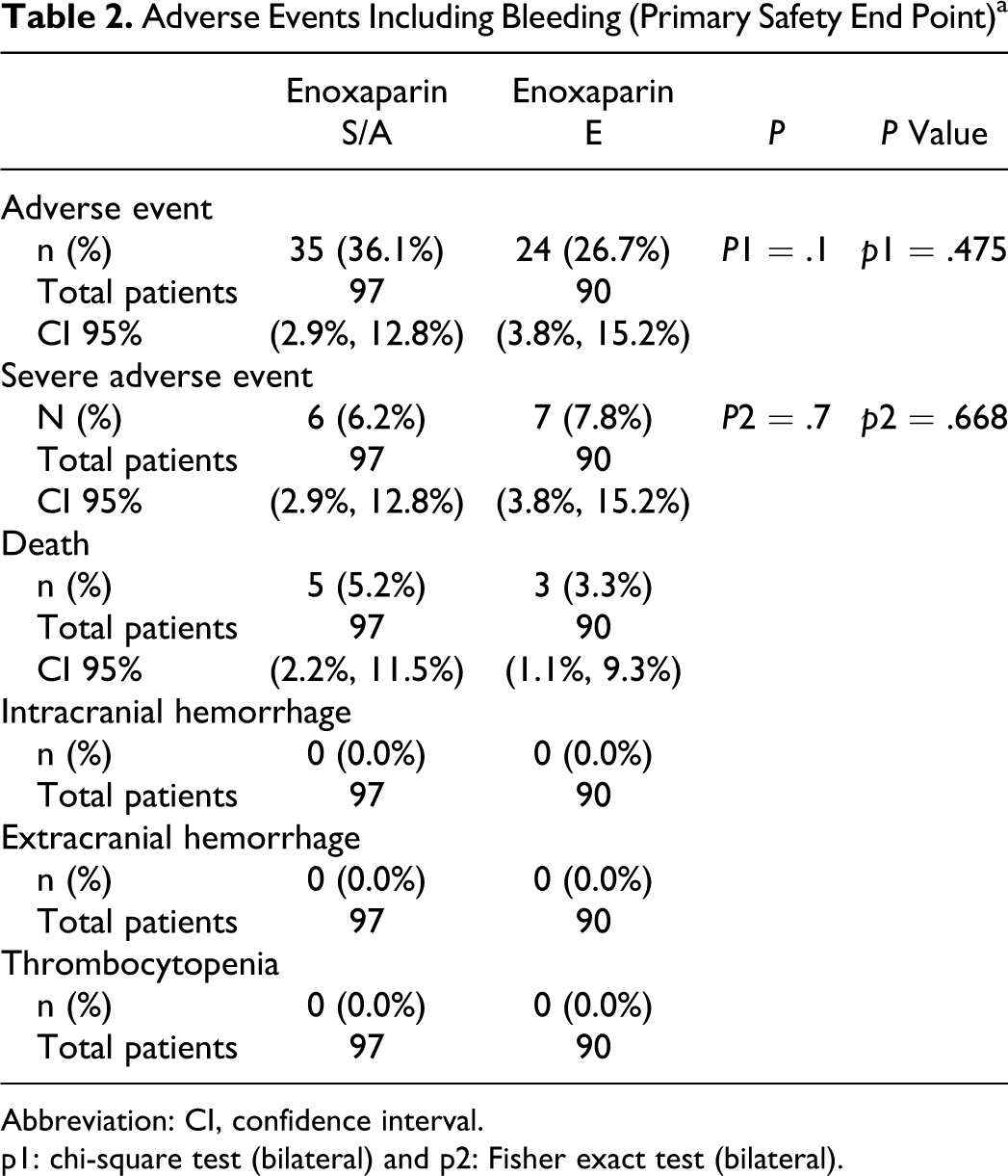
Abbreviation: CI, confidence interval.
p1: chi-square test (bilateral) and p2: Fisher exact test (bilateral).
Signs and Symptoms of VTE (Efficacy Primary End Point)
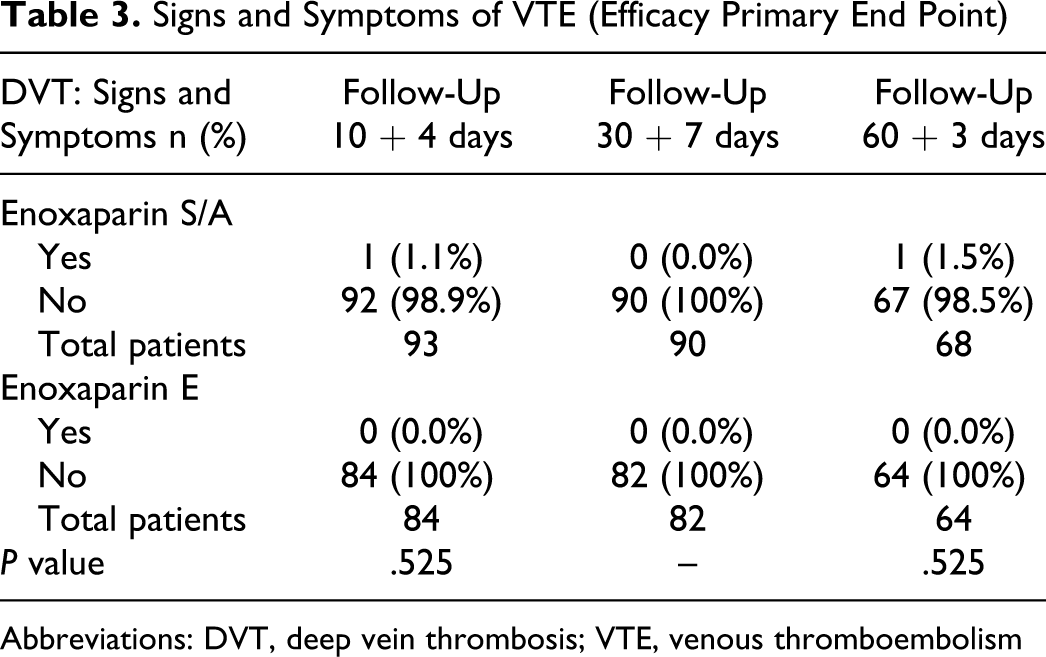
Abbreviations: DVT, deep vein thrombosis; VTE, venous thromboembolism
Minor hemorrhagic events were observed in both groups: in the SAe group these consisted of 1 patient developing abdominal wall ecchymosis, 2 patients developed hematomas of abdominal wall, and 1 episode of hematuria. In the Ee group, 2 patients had flank ecchymosis and 1 had an abdominal wall hematoma. No statistical differences among minor bleedings events were observed (Table 2).
No differences in laboratory evaluations were found. In SAe group hemoglobin (Hb) = 13.4 ± 1.9 g/dL; hematocrit (Hct) = 40.0 ± 5.3%; platelet count = 264 701.0 ± 83 527.2/mm3; activated partial thromboplastin time (aPTT)= 30.8 ± 5.6 seconds; prothrombin time (PT) = 13.1 ± 2.8 seconds; international normalized ratio (INR) = 1.06 ± 0.13; serum creatinine = 0.9 ± 0.2 mg/dL; glutamic–oxaloacetic transaminase (GOT) = 27.9 ± 17.5 U/L, and glutamic–pyruvate transaminase (GPT) = 32.6 ± 23.6 U/L. Similar values were observed in the Ee group: Hb = 13.3 ± 1.7 g/dL; Hct = 39.8 ± 4.8%; platelet count = 262 033.3 ± 88 301.0/mm3; aPTT = 31.1 ± 5.5 seconds; PT = 16.7 ± 19.2 seconds; INR = 1.08 ± 0.14; serum creatinine = 0.9 ± 0.3 mg/dL; GOT = 29.4 ± 29.6 U/L, and GPT = 38.6 ± 51.6 U/L. No thrombocytopenia was observed in any patient enrolled on this trial.
In the SAe group, 6 patients experienced severe adverse events (1 had bronchoaspiration, 1 had wound dehiscence, 2 reoperations for abscess drainage, 1 died due to underlying disease process, and 1 had pneumonia). In the Ee group, 11 patients experienced severe adverse events (subphrenic abscess, enteric fistulae, acute renal failure, acute pancreatitis, 3 reoperations, respiratory failure, and death).
The Anti-factor Xa activity were collected from 20 patients on days 1, 3, and 7, the mean values for each compound were SAe = 0.226 (IU/mL) and Ee = 0.301(IU/mL). No statistical differences were observed on days 1 and 7. However, on day 3, there was a statistical difference in favor of the generic LMWH (SAe = 0.187 IU/mL, Ee = 0.321, and P = .024). No traces of OSCS were found on the batches of enoxaparin, either generic or branded. anti-factor IIa activity of the generic compound was 26.8 U anti-factor IIa/mg, with an anti-factor Xa/anti-factor IIa ratio of 4, posttrial evaluation.
Discussion
The principal action of heparin is to potentiate the antithrombotic effects of antithrombin III. 2,6,8-10 Its primary action is to neutralize activated factor X in the final common pathway of coagulation. The ability of heparin and LMWHs to potentiate the activity of antithrombin III suggests that they have a well-established role in surgery as prophylaxis of VTE. 6 The use of a dose, lower than the dose used therapeutically, lowers the risk of hemorrhage. All these conclusions are well defined with branded LMWHs. 6,8,9,11
The anticoagulant activity of LMWHs is typically assessed according to their ex vivo anti-factor Xa activity. However, Anti-factor Xa activity is not a good predictor of clinical effect evaluation, regarding efficacy (occurrence of DVT/PE) and safety (hemorrhage). Therefore, anti-factor Xa activity should not be extrapolated to clinical practice. The literature widely provided the information that there is no correlation between anti-factor Xa activity and clinical outcome. 1,4,12 New guidelines regarding the use of generic LMWHs are now under development. 13
The trend to use only pharmacokinetic and pharmacodynamic parameters for regulatory approval is spreading worldwide including large countries like China. 14 As the primary aim of a generic drug is to reduce costs without compromising patient care, it is expected that thorough studies are performed before regulatory approval. 1,2,8,10-12,15,16 Because LMWHs represent biological drugs, clinical trials should be used to determine generic equivalency to the branded drug.
To the date, we have found only 1 head-to-head clinical trial on the literature comparing generic versus branded generic LMWH (a pilot study that included prophylaxis and treatment of patients with VTE comparing branded enoxaparin versus Cutenox, also a generic version of enoxaparin). This study showed no differences among compounds. 17
We decided to use major abdominal surgical patients, because lower dose of enoxaparin (40 mg od) for a short period of time (7-10 days), according to the ACCP guidelines, is sufficient to provide acceptable protection against DVT/PE, with low risk of hemorrhage. If signs of complications were observed during this exploratory open-label trial, the study could be interrupted. Moreover, all principal investigators were abdominal surgeons with experience in this study population. The present study shows that comparing patients receiving either the branded enoxaparin or the testing generic version, the od administration of a lower fixed dose of 40 mg (independent of body weight) is equally safe and effective for prophylaxis of VTE. The generic LMWH did not increase the risk of postoperative thrombotic disease (zero DVT/PEs on the tested drug vs 2 DVTs on the branded group with no statistical difference). Those observations persisted at the 2-month follow-up. Importantly, there were no major bleeding events with the new generic version of LMWH compared with the branded one. Minor bleeding rates were comparable in both groups with no statistical differences.
The findings correspond to the expected outcome using low-dose LMWH for VTE prophylaxis. 18,19 However, total incidence of thromboembolism including subclinical cases (often diagnosed by venograms) is obviously much higher than the clinical incidence shown in this study. We decided to use the CUS rather than venograms, because it is a noninvasive procedure, to evaluate the end points. In addition, it is the modality most often used in practice to diagnose DVT. Our major concern, in this very first trial with generic LMWHs in the surgical setting, was safety.
It was observed the generic LMWH presented higher levels of anti-factor Xa activities on day 3, with statistical significance (P = .024), when compared to the branded drug. In addition, an anti-factor Xa/anti-factor IIa ratio of 4 was observed. As expected, no traces of OSCS were found on the samples analyzed. 20
The strengths of our study include its originality (first study with a head-to-head comparison between branded and generic enoxaparin with regard to outcomes in major abdominal surgery) and the homogeneity of the groups evaluated, allowing precise comparisons between both compounds. In addition, despite highlighting major bleeding for statistical analysis, all bleeding events were recorded, and differences in clinically relevant nonmajor bleeding (CRNM) or minor bleeding did not occur. Recently, some medical societies as well as regulatory authorities have published recommendations for the development of generic LMWH. Clinical trials including VTE prophylaxis in high-risk patients or arterial thromboembolism with efficacy and safety end points are common recommendations to all documents, from European Medicines Agency (EMEA), to International Society on Thrombosis & Haemostasis (ISTH) and South Asian Society on Atherosclerosis and Thrombosis (SASAT).
13,21
Our study is the first to address those recommendations. There are some weaknesses in the study. The sample size is small to draw definitive conclusions. Bleeding rates as well as VTE events were below the expected levels. Results of the present pilot study argue for a more detailed appraisal, in larger, randomized controlled trials of those new compounds. Other head-to-head studies compiling biochemical comparative evaluations like thrombin-activatable fibrinolysis (TAFI), tissue factor pathway inhibitor (TFPI) release,
Conclusions
This initial exploratory trial indicates that 40 mg od dose of Ee, the generic LMWH, appears to be as safe and as effective and well-tolerated as the branded enoxaparin (Lovenox or Clexane in Brazil) in the prophylaxis of VTE in such high-risk (major abdominal surgery) patients. A significant higher anti-factor Xa activity of the generic compound was observed on day 3 of treatment with no effect on outcome. Further studies in different indications are widely warranted.
Footnotes
Appendix A
Acknowledgments
The author acknowledge the contribution of the engineer Fernando Pereira Gomes (in memorium), for all the logistics of this multicenter trial; Livia Gomes (study nurse), Ariane Gomes (project manager), Mariana Bergamo (study manager), Alessandro Soares (study monitor), and Eliana Yamashita (co-investigator); and the 4 enrolling sites for their compromise in the enrollment. This study was sponsored by Eurofarma Laboratories. The Authors declare that the sponsor had no involvement in the study design, in the data collection, analysis and interpretation of data, in the writing of the manuscript, and in the decision to submit the manuscript for publication.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed the receipt of the following financial support for the research, authorship and/ or publication of this article: The study was funded by Eurofarma laboratories. The authors received no financial support for authorship and/or publication of this article.