
Abstract
Syncope refers to the transient loss of consciousness and the most common type of syncope is vasovagal syncope (VVS), usually occurring when a person is in an upright position, and/or after exposure to intense stress. The sequelae of VVS is caused by an increase in sympathetic tone and heart rate combined with an underfilled left ventricular chamber that leads to stimulation of cardiac afferent C fibers ultimately leading to bradycardia and vasodilation causing a reduction in venous return, cerebral hypoperfusion and VVS. Several treatment options have been tested including physical counter-pressure measures, electrical pacing, cardioneuroblation. Pharmacological interventions and clinical trials for VVS are summarized in this review; however, there is still limited evidence of their efficacy for reducing episodes of VVS. This review will examine studies using animal models of the vasovagal reflex arc to investigate the physiological mechanisms and neurotransmitters associated with VVS, the tilt-table test that induces VVS in patients and the potential sources of cardiac and platelet mediators that can activate cardiac afferent C fibers. This study will also consider how the previously investigated pharmacotherapies provide insight into the multiple mechanisms involved in VVS and propose new targets for the pharmacological treatment of VVS.
Introduction
Syncope is a syndrome characterized by a transient loss of consciousness, an inability to remain in posture followed by spontaneous recovery. It is crucial to distinguish between a loss of consciousness caused by syncope and other causes including seizures. 1 Syncope can then be further classified into four different categories including reflex (neurally mediated) syncope, cardiac causes, orthostatic hypotension, and unexplained. 2 Reflex syncope is caused by a reflex arc, that results in vasodilation or bradycardia or both. Reflex syncope encompasses three categories which include vasovagal syncope (VVS), carotid sinus syndrome and situational syncope. 3 VVS is the most common form of syncope syndrome. By the age of 60, 42% of women and 32% of men would have experienced at least one episode of VVS in their lifetime. Characteristics of VVS include the following elements: (a) occurs in an upright posture or after exposure to emotional stress, pain, or medical settings; (b) features diaphoresis, warmth, nausea, and pallor; (c) is characterized by hypotension and relative bradycardia; and (d) followed by symptoms of fatigue. 4
Clinical Manifestation of VVS
VVS usually occurs when standing in an upright posture rather than a sitting position. The symptoms that may precede an episode of VVS include sweating, lightheadedness, feeling hot or cold, nausea, palpitations, pallor, dimming of vision or diminution in hearing. 5 In patients experiencing an episode of VVS, the prodromal symptoms may occur for several minutes. Patients may also experience these symptoms with no eventual loss of consciousness, otherwise termed as presyncope. In some instances, patients do not experience any of these symptoms, however these episodes of syncope would be more commonly associated with cardiac causes of syncope. With episodes of VVS, recovery occurs within minutes with a return to normal blood pressure and heart rate. 1
Initial Evaluation
To formulate a diagnostic plan, patients who suffer from syncope are categorized into three groups: those with a cardiac cause, those with a non-cardiac cause, and those with an unexplained origin. This must be rapidly established as patients with a syncope caused by cardiac problems are at a relatively high risk of sudden death. VVS is a non-cardiac cause of syncope. 1 The initial evaluation of patients with suspected syncope is reliant on the age of the patient with the aim of excluding cardiac disease. Elderly patients have a higher priority for screening as they are at a higher risk of experiencing a cardiac cause syncope. Assessments may include the use of a carotid sinus massage and cardiac testing. In all patients experiencing suspected episodes of VVS, the initial evaluation includes reviewing their medical history, a physical examination and electrocardiography (ECG). 6
History and Physical Examination
Diagnosis of VVS is primarily based on the clinical features of the event.
7
History taking is a crucial step in determining whether fainting has occurred due to an episode of VVS or other causes. Medical history includes providing details of predisposing situations, presence of prodromal symptoms, frequency and duration of episodes, recovery time and symptoms after syncope, medication use, family history of syncope or sudden unexplained death, comorbidities and a history of preexisting conditions, particularly cardiovascular disease.
3
The following characteristics are associated with, but not diagnostic of, non-cardiac cause syncope, especially VVS
3
:
Younger age No known cardiac disease Syncope occurs after orthostatic stress Presence of prodromal symptoms of VVS Presence of triggers, such as pain and intense stimulus A prolonged history of similar episodes of syncope
A physical examination would exclude a susceptibility to orthostatic hypotension and any cardiac and/or neurological findings. A slow, rapid, or irregular heart rate may be detected due to rhythm disturbances, atrial fibrillation, atrial flutter or ectopy which may indicate structural heart disease and should be confirmed by an ECG as patients with VVS often have normal ECGs. 3 Other factors include the potential for physical injury and whether the prescribed treatments require hospital monitoring. With VVS, the risk of short-term death is low, however, there may be risks associated with injury upon falling.
Diagnosis of VVS
The diagnosis of VVS may involve the tilt-table test, a procedure that involves a patient lying down on a table and the table being tilted from a horizontal position to a vertical position (to 70 degrees) to simulate standing up. Heart rate and blood pressure changes are monitored as the patient's position changes into an upright position. The tilt-table test creates a gravity dependent VVS, the movement to an upright posture leads to blood pooling, central volume depletion and reduced cardiac preload. Baroreceptor mediated increases in sympathetic nervous system output to maintain blood pressure causes an increase in heart rate. Increased cardiac contractility of underfilled ventricles activates the cardiac mechanoreceptors, the activation of C afferent vasovagal pathways to the medullary region of the brain, leading to an increase in efferent parasympathetic outflow and withdrawal of sympathetic tone. VVS patients then experience bradycardia or phases of asystole and/or vasodilation or venodilation. 8 The tilt-table test is an effective diagnostic tool for VVS for several reasons. Firstly, both induced and spontaneous VVS episodes are characterized by similar prodromal symptoms and signs, such as nausea and pallor. 9 Secondly, the changes in blood pressure and heart rate during a tilt-table test mimic those in a VVS episode. 10 Thirdly, increased levels of plasma catecholamines have been reported in both tilt-table testing and spontaneous VVS.11,12 Both noradrenaline and adrenaline levels increase through the tilt-table test, however significant increases in adrenaline only have been observed in patients exhibiting tilt-table induced VVS compared to those who were negative for the tilt-table test.12,13 A positive result in a tilt-table test is one where symptoms of syncope caused by hypotension or/and bradycardia occur as a result of the procedure and mimic the patient's usual syncopal episodes. Heart rate or blood pressure changes alone do not account for a positive result. 9 A positive test establishes a patient's tendency to VVS rather than establishing the cause of syncope. 4 Several pharmacological agents can compromise the specificity of the test. High doses of isoprenaline, for example, during a tilt-table test exceeding 10 minutes at 80° tilt upright reportedly reduced the specificity of the procedure. 14 The tilt-table test requires further investigation as to its accuracy in evaluating VVS as protocols for this use of the test varies between clinics. 4 The tilt-table test is helpful in circumstances where fainting occurs in a high-risk setting and in the absence of organic heart disease or in the presence of organic heart disease with evidence suggesting vasovagal episodes. 4
The Physiology of the Vasovagal Reflex
The physiology of an episode of VVS is not fully understood but involves the activation of cardiac C fibers. Triggers such as intense emotion, pain, or the sight of blood can cause an increase in sympathetic tone, leading to an increase in heart rate and contractility and stimulation of mechanosensitive cardiac C fibers, 15 see Figure 1. Research into circulating catecholamine levels associated with an episode of VVS consistently report a relatively early increase in circulating adrenaline during head-up posture prior to the faint. 16 Activation of afferent cardiac C fibers leads to stimulation of the medullary vasodepressor region in the brain, which activates increased efferent vagal outflow to the heart to cause bradycardia and inhibits vascular sympathetic tone to induce vasodilation. Cerebral perfusion is highly dependent on systematic arterial pressure, which is influenced by cardiac output and total peripheral vascular resistance. Therefore, a decrease in cardiac output decreases systematic arterial pressure resulting in cerebral hypoperfusion which causes the symptoms associated with episodes of VVS.15,17

Physiological processes that result in VVS episodes.
Receptors Coupled to Afferent C Fibers in Cardiac Vagal Nerves
Cardiac receptors associated with the afferent nerve fibers within the cardiac vagal nerves are connected to two major fiber types. 18 Receptors located in the walls of the atria and veno-atrial junctions are linked to medullated fibers with conduction velocities which range from 8 to 32 m s−1. The other receptor type, which appears as the terminal portion of a network of non-medullated fibers are widely distributed in the walls of all cardiac chambers. As the conduction velocity of these fibers is less than 2.5 m s−1, they are classified as C fibers. 18 Receptors for the non-medullated cardiac vagal afferents are widely distributed throughout the heart including intra-arterial and internal ventricular septa. During resting conditions these receptors are silent or have a sparse irregular discharge rate of <1 mp s−1 and most of these receptors discharge in sequence with cardiac rhythm. Increases in receptor discharge occurs with an increase in atrial and ventricular end diastolic pressure following increases in atrial and ventricular pressure caused by transfusion or by outflow occlusion. 18 The discharge of ventricular receptors also increases with the administration of positive inotropic drugs such as isoprenaline, hence the activation of left ventricular receptors associated with non-medullated afferents is caused by increases in pre-load and after-load and changes in cardiac contractility. 19 In experiments using anesthetized atropinized dogs, the afferent vagal traffic from cardiopulmonary receptors was prevented through sectioning of aortic nerves and denervation of the carotid sinuses, causing arterial hypertension, increased renal and mesenteric vascular resistance and tachycardia.20,21 These observed changes were caused by an increase in sympathetic adrenergic outflow with no involvement of the sympathetic cholinergic nerves. These experiments demonstrated the dominant role of vagally innovated cardiopulmonary receptors and that their inhibition of the vasomotor center is as effective as the arterial baroreceptors. Vagal afferent C fibers are important contributors to neurogenic cardiovascular regulation. As previously discussed, receptors attached to these fibers are activated by changes in cardiac pressure, however, they have been reported to also be activated by chemical stimuli.
Chemical Stimulation of Cardiac Vagal Afferent C Fibers
The proposed pathophysiology of VVS is that it is initiated by specialized cardiac myocardial pressure sensitive and chemo sensitive receptors located in the left ventricle. 22 The serotonin 5HT3 receptor, the transient receptor potential vanilloid 1 (TRPV1) receptor and the purine P2X2/3 receptor are the three main types of ion channel coupled receptors that have been identified, that can activate cardiac afferent vagal C fibers. In the anesthetized rat model, the cardiac vagal afferent C fibers were reported to be activated by increased mechanical pressure or serotonin via the serotonin 5HT3 receptor 23 and considered to be bimodal. Gene expression and receptor binding studies have established a presence of the serotonin 5HT3 receptors on the rat vagal nerve fiberes.24,25 Another chemical substance known to activate cardiac vagal afferent C fibers is capsaicin, a selective agonist of the vanilloid TRPV1 receptor. When another vanilloid TRPV1 receptor agonist cinnamaldehyde was injected intravenously into anesthetized mice, a transient hypotensive response with decreased heart rate was observed. 26 Hence the vanilloid TRPV1 receptor has been identified as another ion channel coupled receptor that mediates bradycardia and hypotension via the cardiac vagal afferent C fibers. 27
The vanilloid TRPV1 receptor is an ion channel located on sensory nerves that is stimulated by heat, protons, capsaicin and other endogenous lipids called endovanilloids 28 and has a role in sensing noxious stimuli to elicit counteractive measures to reduce pain and/or injury. As such, significant increases in heart rate and contractility observed at the start of an episode of VVS may activate vanilloid TRPV1 receptors on cardiac vagal afferent C fibers in a reflex response to slow the heart as a cardioprotective measure.
Adenosine triphosphate (ATP) has also been reported to activate the vagal depressor reflex response in the heart. 29 Extracellular ATP regulates the cardiovascular system through activation of cell surface receptors. 30 There are two purine P2 receptors subtypes that have been identified; P2× receptors which are coupled to ion channels and the P2Y receptors which are G protein coupled receptors. The half-life of extracellular ATP is very short as it is catabolized rapidly to ADP, AMP and adenosine. 31 ATP can activate vagal sensory nerve terminals located on the left ventricle, triggering the central vagal depressive reflex. The role of ATP in activating the vagal depressor reflex was studied in a closed chest canine model where ATP was administered via the right coronary artery and left circumflex coronary artery. 32 ATP exerted a negative chronotropic effect on sinus node automaticity. The effect of ATP was completely abolished by the intravenous muscarinic receptor blocker atropine or intra-left circumflex coronary artery application of a potent purine P2X2/3 receptor antagonist 2′,3′-O-(24,6-trinitrophenyl)-ATP (TNP-ATP), but not potent purine P2X1 or P2X3 receptor blockers. 32 Xu et al 32 concluded that ATP activates a cardio-cardiac vagal depressor reflex through stimulation of purine P2X2/3 receptors located on vagal sensory terminals located in the left ventricle. Finally, acute myocardial ischemia, which causes platelet activation and the release of metabolites such as serotonin, protons and anandamide, was shown to enhance the vanilloid TRPV1 and serotonin 5-HT3 receptor medicated Bezold-Jarisch reflex in rats. 33
Cardiac Sources of Serotonin and ATP
There are multiple sources of serotonin and ATP in the heart including platelets, where these compounds contribute to physiological and pathophysiological roles in cardiac function and under appropriate conditions may activate receptors in the cardiac afferent C fibers, see Figure 2. Approximately 90% of serotonin is present in intestinal enterochromaffin cells, 5% in platelets and 2% in the brain. 34 Circulating platelets in the intestine take on serotonin through the serotonin transporter (SERT) where it is stored in the dense granules with calcium and ATP. Serotonin, is released in the heart from platelets during pathophysiological conditions such as coronary endothelial damage or stenosis. 35 Serotonin has been detected in both mouse and human cardiac tissue as well as in rat neonatal cardiomyocytes and has several effects on the cardiovascular system including increased heart rate and force of contraction, fibrosis of cardiac valves, coronary vasoconstriction, arrhythmias and thrombosis. 36

Schematic diagram of potential sources of catecholamine/purinergic/serotonergic activation of VVS. ATP, adenosine triphosphate; ADP, adenosine diphosphate; AMP, adenosine monophosphate; AD, adenosine; ADR, adrenaline from adrenal medulla; NA, noradrenaline; 5-HT, serotonin; +, activation of platelets; –, inhibition of platelets; 5-HT2A, serotonin 5-HT2A receptor; 5-HT3, serotonin 5-HT3 receptor; α2-ADR, α2-adrenoceptor; β2/3-ADR, β2- or β3-adrenoceptor; P2X2/3, purine P2X2/3 receptor; P2Y1, purine P2Y1 receptor; P2Y12, purine P2Y12 receptor; TRPV1, transient receptor potential vanilloid 1 receptor.
There are multiple sources of ATP release in the heart. Firstly, ATP is co-released with noradrenaline from sympathetic nerves that innervate the heart. 31 Secondly, red blood cells release ATP from their cytosolic pool of ATP in response to conditions such as hypoxia, 37 deformation 38 and sheer stress 39 via Pannexin 1, a protein hemichannel for ATP release.40,41 ATP release from red blood cells can also be stimulated by prostacyclin released by endothelial cells subjected to shear stress, through a voltage dependent anion channel.42,43 Thirdly, ATP has been observed to be released from ischemic myocytes to cause an atropine-sensitive bradyarrhythmia associated with a myocardial infarction. 44 Finally, activated platelets release the adenine compounds ATP and ADP stored in the dense granules following blood vessel injury, reaching physiological levels of 20 µM 45 or 50 µM 46 respectively in the microenvironment. Agonists known to activate platelets and cause the exocytotic release of ATP/ADP include thrombin, adrenaline, collagen and ADP.
A Role for Catecholamines in Precipitating the VVS Reflex
During a response to stress, an elevation in the catecholamines noradrenaline and adrenaline activate the physiological processes that ultimately initiates the VVS reflex. Catecholamine levels have been shown to rise significantly during the tilt-table test in studies performed by Sander-Jensen et al. 12 Noradrenaline released from cardiac sympathetic nerves and adrenaline from the adrenal medulla cause an increase in heart rate observed during the tilt-table test. A characteristic of individuals who exhibit VVS is the drop in central venous pressure and pulse pressure, possibly due to activation of β2-adrenoceptors by adrenaline, which is more potent at vascular β2-adrenoceptors then noradrenaline.47,48 The increase in heart rate with a reduction in central venous pressure may increase activation of the ventricular mechanoreceptors coupled to the vagal C-fiber afferents. As the plasma levels of adrenaline continue to rise prior to the development of syncope in VVS patients,12,49 adrenaline may also activate platelet adrenoceptors and in combination with locally released mediators in the heart, stimulate the release of serotonin and ATP from platelets (see Figure 2).
A Role for Platelets in Activating the VVS Reflex
The rise in adrenaline that occurs during the sequelae of VVS may work as a co-activator of platelets with ATP/ADP or serotonin. Adrenoceptors are located on mammalian platelets although the relative activity of the adrenoceptors varies according to species. 50 With respect to human platelets, adrenaline can stimulate an aggregatory response through α2A-adrenoceptors 51 while activation of β2-adrenoceptors inhibits platelet function. 52 However, more recently the α2B-adrenoceptors (pro-aggregatory) and the β3-adrenoceptors (inhibitory) have also been identified in human platelets.53,54 Adrenaline is considered a weak platelet agonist whose function is to sensitize the platelets to other activating agents.55,56 Adrenaline potentiates ADP induced platelet aggregation, an effect that was reversed by the α2-adrenoceptor antagonist yohimbine. 57 ADP and ATP are stored within the dense (δ) granules of platelets with serotonin. 57 ADP can induce platelet aggregation through the two purinergic P2Y1 and P2Y12 receptors 31 and can cause reversible platelet shape change, integrin αIIbβ3 activation, and fibrinogen binding. 58 ATP alone does not stimulate platelet aggregation but in combination with noradrenaline or adrenaline can activate platelet aggregation. 59 ADP (either as a breakdown product of locally released ATP or from platelets) causes a biphasic aggregation response with the release of serotonin to activate serotonin 5-HT2A receptors. Binding of serotonin to 5-HT2A receptors triggers classical Gαq-linked signal transduction events through the PLC pathway resulting in calcium mobilization and initiating multiple effects including platelet shape change and the activation of small GTPases, which promotes the exocytosis of granules from the platelet. Similar to adrenaline or ATP, serotonin is primarily regarded as an amplifier of platelet activation events as it only stimulates small increases in intracellular calcium levels and alone is not sufficient to induce platelet aggregation, 60 but can potentiate aggregation with other stimuli. 61 The free circulating level of serotonin in plasma is kept between 15 and 120 nM and it has to reach micromolar levels at sites of injury to activate platelets. 62 It has been demonstrated that serotonin alone does not stimulate platelet aggregation but potentiated a pro-aggregatory response to adrenaline. The synergistic interaction of serotonin and adrenaline is inhibited by the α2-adrenoceptor blocker yohimbine and calcium channel blockers verapamil and diltiazem. 61 For a summary of ligands that activate platelet and are released from platelets see Figure 2.
The Role of Serotonin in Blood Pressure Control
The hypotensive and bradycardic responses observed during an episode of VVS, is due to withdrawal of sympathetic control of the cardiovascular system, or a failure of the autonomic nervous system to adequately compensate for physiological responses to stress. This mechanism is similar to that observed during a hemorrhage. The role of the serotonergic system in blood pressure responses during a hemorrhagic episode can mimic that observed during VVS. One study has observed a relationship between endogenous opioid receptors and central serotonergic pathways as the hypotensive effect of β-endorphins is attenuated by pretreatment with serotonin receptor antagonists or inhibitors of serotonin production in cats. 63 Another study showed that in dogs, when introduced into the cerebroventricular system, serotonin agonists can induce hypotension. 64 These studies suggest that anti-serotonergic agents can reduce depression of the cardiovascular system which occurs following acute blood loss. To test this theory, methysergide a serotonin blocker and p-chlorophenylalanine (PCPA), an inhibitor of tryptophan 5-hydroxylase were tested in the treatment of acute hemorrhagic shock in cats. 65 The mean arterial pressure of cats that were treated with PCPA was 109 ± 6 mm Hg compared to 125 ± 9 mm Hg for the placebo group, which was not statistically different. However, by the end of the hemorrhage, the blood pressure of cats in the placebo group was significantly lower (36 ± 5 mm Hg), as opposed to 83 ± 7 mm Hg in the PCPA pretreated group. After 2 hours of recovery, the blood pressure increased in both groups to 70 ± 7 mm Hg in the placebo group and 83 ± 7 mm Hg in the PCPA pretreated group. The bradycardic responses observed in the PCPA group pretreated group were significantly lower than that in the placebo group. The use of methysergide treatment in acute hemorrhagic shock in cats showed a significant difference between the MAP in the methysergide treated group at the end of the experiment (102 ± 5 mm Hg), and that of the control group (70 ± 7 mm Hg). There were no changes in the heart rate in the two groups. Methysergide has also been observed to increase arterial pressure in conscious rabbits who experienced hypotension due to experimental hemorrhage. 66 Hemorrhage causes the activation of sympathoinhibitory serotonergic mechanisms in the CNS, which then results in bradycardia and vasodilation. 66 Another study investigated the role of central serotonergic neurotransmitters in neurocardiogenic syncope using twenty participants (twelve of whom tested positive for a tilt-table test, and eight of whom tested negative for a tilt-table test) who were injected with clomipramine, a serotonin reuptake inhibitor. 67 Plasma prolactin and cortisol levels were then measured as the secretion of prolactin and cortisol is regulated by serotonin, and thus, an increase in prolactin and cortisol was said to be attributed to the activation of the central serotonergic system. The study showed that patients with a history of VVS, have a different hormonal response to the serotonin reuptake inhibitors than patients with no history of VVS, as significant increases in prolactin and cortisol were observed in the former group. This suggests that the activation of the serotonergic system is involved in the pathogenesis of VVS. Other studies using dogs have reported contradictory results regarding the role of serotonin in regulating blood pressure. One study reported that an intercerebroventricular injection of serotonin (0.25-5 mg) in anesthetized dogs with excised vagus-sympathetic-depressor trunks lead to a fall in blood pressure that persisted for 2 hours. 68 The usual pressor response to bilateral carotid occlusion, as well as sympathetic activity, was also reduced. However, in another study, 5-hydroxytryptophan (5-HTP) appeared to have no effect on blood pressure except when dogs were pretreated with peripheral decarboxylase and monoamine oxidase inhibitors 69 while another study using conscious dogs showed that 30 mg/kg 5-HTP increases blood pressure. 70 Circulating levels of free serotonin are high in animals with hypertension further suggesting the role of serotonin in increasing blood pressure. 71
Peripheral serotonin 5-HT3 receptors have been linked to VVS. In two animal studies, inhibition of serotonin 5-HT3 receptors prevented components of VVS reflex that were observed following the administration of serotonin. A pilot study investigated the effect of serotonin subsequent to pretreatment with granisetron, a serotonin 5-HT3 receptor antagonist, and found that blood pressure was maintained in the anesthetized rat. 72 Another study using a hemorrhagic model of syncope reported that granisetron inhibited bradycardia and maintained blood pressure in the rabbit model of the Bezold-Jarisch reflex. 73 Ondansetron 4 mg/10 ml saline IV, was used to prevent the Bezold-Jarisch reflex in elderly patients undergoing general anesthesia in a randomized double-blind placebo controlled trial, however ondansetron was effective in preventing post-induction hypotension but not bradycardia. 74 A pilot study was also performed in 8 patients to test the efficacy of 160 µg/kg granisetron in preventing VVS during tilt-table testing and found that 50% of patients had a negative-tilt test. 75
Central Control of Vagal Inputs into the Heart
Cardiac vagal preganglionic neurones are located in the dorsal vagal nucleus and the nucleus ambiguous. Serotonin is released at multiple sites that control autonomic outflow including the nucleus tractus solitarius (NTS) where cardiorespiratory afferent fibers terminate and the cardiac vagal preganglionic neurones and rostral vasolateral medulla, the location of the sympathetic premotor neurones. Several serotonin 5-HT receptor subtypes have been identified in brainstem regions that are involved in cardiac control, with serotonin 5-HT1A, 5-HT3 and 5-HT7 receptors specifically implicated in cardiovascular reflexes.76,77 Inhibition of serotonin 5-HT1A receptors attenuates bradycardia caused by stimulating baroreceptor and cardiopulmonary afferents but not chemoreceptors and are located in the rostral vasolateral medulla. 78 Whereas inhibition of serotonin 5-HT7 receptors in the NTS inhibits all reflex bradycardias. 76 Blocking serotonin 5-HT3 receptors reduce cardiopulmonary reflex induced bradycardias. The bulk of the serotonin 5-HT3 receptors are located on primary afferent terminals to elicit vagal bradycardia where activation of serotonin 5-HT3 receptors excite cardiac vagal preganglionic neurones and the NTS via a glutamate dependent mechanism.76,77
Purine receptors also have a central role in the VVS reflex. The NTS integrates visceral afferent reflexes and P2X2 and P2X3 receptors have been reported in the NTS of rats. 79 P2X7 receptors have been located presynaptically on vagal afferents in the NTS and injection of α,β-meATP into the NTS causes bradycardia and hypotension. 80
Options for Tilt Table Testing
The tilt-table test was first proposed as a diagnostic tool for suspected VVS in 1986. 81 The use of tilt-table testing was originally implemented to investigate the pathophysiological responses to postural changes, and previous clinical studies later have shown that the tilt-table test does not provide prognostic predictive power or has been shown to be useful in selecting effective therapies for VVS. 82 However, a positive tilt-table test is currently used to select patients for a CLS-pacemaker following a study showing CLS pacing significantly reduced syncope burden and time to first recurrence by 7-fold, prolonging time to first syncope recurrence in patients age ≥40 years with head-up tilt test-induced vasovagal syncope compared with sham pacing. 83 As we learn more about the triggers, receptor contributions and reflex pathways, the choice of medications tested previously in clinical trials are selected for specific mechanisms may actually be causing VVS episodes due to off target effects. For example, clomipramine a tricyclic antidepressant has been shown to have moderate effects increasing the rates of VVS in tilt-table positive patients 84 and was implemented to alter central levels of serotonin via inhibition of the serotonin transporter. However, tricyclic antidepressants are well established to block α1-adrenoceptors to cause orthostatic hypotension and nonselective inhibition of acetylcholine muscarinic receptors to induce sinus tachycardia which in combination are known precipitants of VVS.
Chemical stimulation of the vasovagal reflex and episodes of VVS from a cardiac source point to serotonin via serotonin 5-HT3 receptors and/or ATP via purine P2X2/3 receptors (see Figure 2). As previously discussed, blocking serotonin 5-HT3 receptors with compounds such as granisetron negate tilt-table test induced VVS in approximately 50% of patients, 75 whereas the remaining patients may be sensitive to purine induced VVS. The administration of adenosine or ATP have been suggested as potentially more useful noninvasive tools for clinical evaluation of patients with newly diagnosed bradycardia related syncope. 85 The use of ATP as a stimulatory agent for the vasovagal response either during tilt-table testing or in the supine position in patients with neurally mediated syncope or syncope of unknown origin 86 may demonstrate whether VVS is purinergic in origin. Brignole et al have previously reported that the tilt-test and ATP test are poorly correlated in stimulating neurally mediated syncope, 87 again suggesting different triggers for VVS that need to identified for each patient. Also, as ATP is rapidly metabolized to adenosine, which induces atrioventricular block, so would need to be tested with a selective adenosine A1 receptor antagonist such as rolofylline. 88 Or alternatively, selective purine P2X2/3 receptor antagonists could be used to identify purine induced VVS, however as this class of drug is currently in phase III trials and not approved for clinical use as yet. Medications that stabilize platelets or inhibit platelet activation may be effective in the meantime.
Treatment of VVS
VVS is benign in nature, however, recurrent episodes of VVS can compromise a patient's lifestyle and expose a patient to injury upon falling or pose great harm to the patient and the community when fainting occurs in high-risk settings. No specific treatment for VVS has been consistently effective in mitigating episodes of VVS. Several treatment options are available, and the choice of treatment depends on age, the severity of the fainting episodes and clinical forms of syncope. The first-line management of VVS is education and lifestyle modifications. This includes education about the benign nature of the disease, education regarding possible triggers and situations that induce VVS, and the recognition of prodromal symptoms in order to take action, such as isometric movements until they can lie down with raised legs. There are no randomized control studies investigating the effect of education and lifestyle modifications on episodes of reflex syncope, however, education and lifestyle modifications have been proven to be very successful in reducing episodes of VVS. 89 An avoidance of agents that lower blood pressure is another measure that can reduce recurrent episodes of syncope and includes antihypertensive drugs, nitrates, diuretics and antipsychotic drugs. 89 In the Systolic Blood Pressure Intervention Trial, patients with high cardiovascular risk who were on antihypertensive drugs targeting a systolic blood pressure of 120 mm Hg had twice the risk of syncope as opposed to the control group which was targeting a systolic blood pressure of 140 mm Hg. 90
Physical counter-pressure maneuvers (PCMs) include isometric muscle contraction that reduce the onset of fainting, such as leg-crossing, leg pumping, and arm tensing. Three clinical trials and one prospective multicenter randomized trial showed that PCM of legs and arms avoided or delayed fainting.91–94 PCM reduces venous pooling in lower extremities thus increasing cardiac output and preventing VVS. However, for patients with no or minimal prodromal symptoms or patients over the age of 60, PCM may not be as useful. 95 If VVS is caused by orthostatic stress, then a treatment option often used is tilt-table training, where patients remain in an upright position for a certain period of time. However, five controlled trials have shown that no significant impact on the recurrence of syncope has occurred with this type of treatment. 89 Permanent cardiac pacemakers have also been used for patients suffering from recurrent VVS. Dual-chamber permanent pacing with a rate-drop response or “closed-loop stimulation” were found to be successful in reducing and eliminating symptoms in patients with syncope caused by a cardioinhibitory response. 96 The rate-drop response provides a high pacing rate when a vasovagal episode is detected, while the closed-loop stimulation assesses cardiac contraction changes and intervenes when the heart rate declines to prevent fainting. However, in patients who experience a fall in blood pressure prior to a decreased heart rate, cardiac pacemakers may not be efficient. 97 According to evidence from clinical trials, pacemaker therapy was only be effective in patients with recurrent VVS and an asystole of greater than or equal to 3 s 98 which has limited its use. More recently, closed loop stimulation pacing has emerged as a potential solution through using a sensing algorithm that can detect alterations in cardiac contractility and a drop in blood pressure to elevate heart rate with multiple studies that have assessed its efficacy demonstrating superiority to conventional pacing. 99 Also, the intervention of cardioneuroablation, which involves denervation of the cardiac vagal nerve plexi has shown success in reducing VVS in patients with refractory VVS in a meta-analysis. 100
If education and lifestyle modifications as well as PCM are not found to be effective, several pharmacological treatments are available. Table 1 shows a list of pharmacological treatments that have been used or trialed to treat VVS, their mode of action, as well as their effectiveness in reducing episodes of syncope according to previous studies.89,101 A psychiatric approach has also been suggested as psychiatric disorders can accompany VVS and symptoms are similar to panic disorder where VVS can occur following a trigger, with or without warning. 102
Pharmacological Treatments That Have Been Used or Trialed for VVS, Their Mode of Action and Their Reported Effectiveness in Treating Syncope.
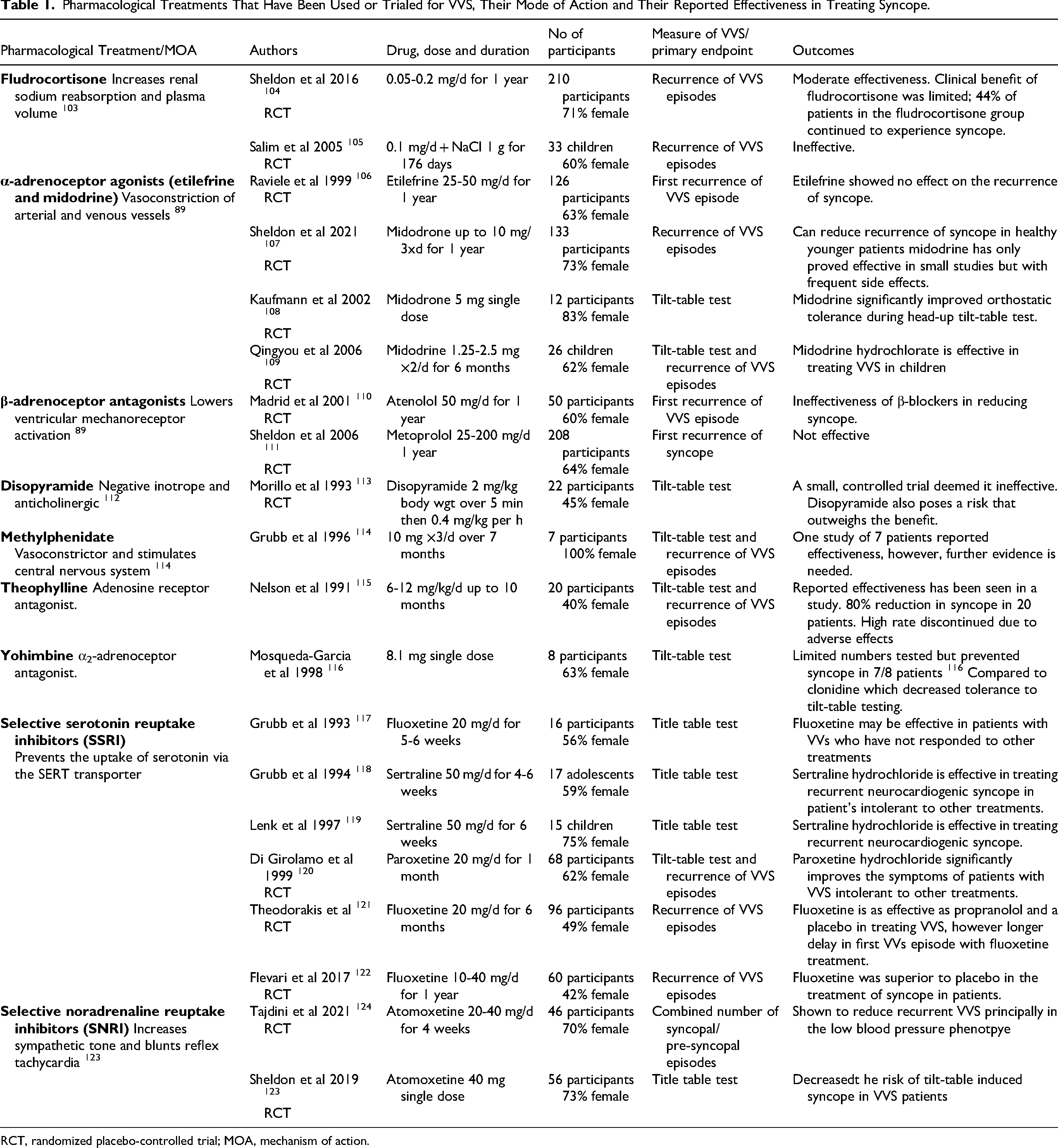
RCT, randomized placebo-controlled trial; MOA, mechanism of action.
The pharmacological interventions for the prevention of VVS are summarized in Table 1. Studies published in the early 1990s were designed as pilot studies, whereas from late 1990s onwards, pharmacotherapies have been tested in randomized placebo-controlled trials where participants are included on the basis of history of VVS episodes and positive tilt-table tests. In general, the pharmacological agents tested were selected on their capacity for symptomatic control of VVS through altering blood volume or vascular or cardiac function. Several recent systematic reviews have been published including a meta-analysis of the use of midodrine, whose mechanism of action is to negate the vasodilator effects of an episode of VVS and was found to be effective in preventing syncope induced by head-up-tilt testing. 125 However, mildrodine has limitations to its wider implementation due to increases in blood pressure, particularly in the elderly cohort. 107 Another systematic review of SNRI inhibition of VVS by agents such as atomoxetine reported that this class of drug is a potential option for the treatment of recurrent syncope. 126 Overall, many of the studies listed in Table 1 demonstrate little to moderate efficacy in reducing tilt-table testing induced syncope or VVS episodes over time in participants. Finally, a systematic review of all the published RCTs confirmed that midodrine was effective at reducing spontaneous syncopal events 127 and the SSRI fluoxetine should be investigated further.
Studies on the Effect of SSRIs in Treating Vasovagal Syncope
As previously discussed, the central serotonergic system is involved in the pathogenesis of VVS. 76 Accordingly, a number of clinical trials investigating the efficacy of SSRIs for treating VVS have been published see Table 1. One study, carried out in 1993, tested the effect of fluoxetine, in the prevention of upright tilt-table induced syncope. Of the patients who became tilt-test negative, 53% showed no symptoms during a follow-up period of 19 ± 9 months suggesting that fluoxetine may be a possible treatment for recurrent VVS. 117 Another study tested the role of sertraline in preventing recurrent VVS in 15 children who had been unresponsive to standard pharmacotherapy with atenolol or disopyramide. 119 Two participants experienced syncope while on therapy and three participants were intolerant to the drug. Among the ten participants that remained, six became tilt-table test negative and reported no symptoms in a follow-up period of 7 ± 3 months, while four stayed tilt-table test positive. This led to the conclusion that sertraline could be a possible treatment for VVS. 119 A larger randomized, double-blind, placebo-controlled study was carried out to test the effect of paroxetine or placebo in patients in whom β-adrenergic blocking agents, vagolytic, negative inotropic or mineral corticoid agents had been ineffective in treating episodes of VVS. The study concluded that paroxetine is effective in reducing VVS episodes with the results showing that 61.8% of participants in the paroxetine group tested negative for a tilt-table test as opposed to 38.2% of participants in the placebo group. In the follow-up period, syncope was reported in 52.9% of patients in the placebo group, and 17.6% of patients in the paroxetine hydrochloride group. 120 Another study tested the effectiveness of sertraline in treating VVS in young patients who were unresponsive to fludrocortisone, transdermal scopolamine, β-adrenergic blocking agents and disopyramide. The study concluded that sertraline is effective in reducing VVS episodes with nine patients out of 14 having a negative tilt-table test. 118 A prospective, randomized, placebo-controlled study compared the therapeutic efficacy of propranolol, a β-blocker which was previously deemed to be ineffective in treating VVS, versus a placebo or fluoxetine. A positive tilt-table test was used to generate an episode of VVS and a questionnaire was used to assess the well-being of participants. The study concluded that all three medications were equally effective in decreasing VVS episodes as no difference was observed in the number of patients who experienced syncope, presyncope or both during therapy. However, on-treatment analysis has shown that fluoxetine is more effective than propranolol and the placebo in increasing the time for the first syncope episode and decreasing the total number of patients who experienced syncope at least once, the number of patients with presyncope, and the mean number of presyncope episodes in the follow-up period. These results were attributed to the fact that fluoxetine improved the quality of life of patients with VVS. The efficacy of fluoxetine cannot be fully proven as a presyncope episode doesn’t equate an episode of VVS. It is possible that the efficacy of fluoxetine was due to its antidepressant action although participants with depression had been excluded. 121 Generally, SSRIs relieved VVS in approximately half the patients tested, indicating that while serotonin may play a significant part in the pathophysiology of VVS, there are other mechanisms involved and differentiating causes of VVS using the tilt-table test with pharmacological interventions may provide a mechanism to identify causes and potential targeted pharmacological therapies for the treatment of VVS. More recently, a study was completed using fluoxetine in patients who test positive on the Anxiety Sensitivity Index and reported that the SSRI was superior to placebo in preventing recurrent VVS. 122 A systematic review and meta-analysis of SSRI inhibition of VVS by Raj and colleagues 128 concluded that this class of drug may be effective in reducing tilt-table induced syncope and recurrent VVS in patients. While the clinical trials are limited in patient numbers, the conclusions of many of these studies do suggest a role for SSRIs in the treatment of VVS and more importantly, points to a role of serotonin the pathogenesis of VVS.
Explaining Previous Pharmacological Clinical Trials from a Platelet Perspective
Currently, there is no direct evidence for activated platelets releasing ligands that stimulate the VVS reflex. However, reviewing previous published clinical trials in the context of platelet physiology may help explain the limited success of some of these trials. The limited choice of pharmacological agents used as therapy for VVS have shown significant effectiveness in large, randomized, controlled trials. 8 However, clinical management of VVS may benefit from further evaluation of the causes of activation of cardiac vasovagal receptors and selecting relevant treatments to decrease symptom burden. Theophylline was implemented in VVS trials to inhibit the actions of adenosine inducing bradycardia and vasodilation. Adenosine, a breakdown product of ATP, has a half-life of 1.5 seconds 129 and is unlikely to produce a sustained depression of the cardiovascular system. PGE1 and theophylline potentiate each other to inhibit ADP-induced aggregation 130 and collagen induced aggregation 131 an effect explained by PGE1 stimulating platelet cyclic AMP formation and theophylline, which inhibits platelet cyclic AMP phosphodiesterase 130 inhibiting its breakdown and reducing platelet activation and aggregation. An alternative drug that is known to stabilize platelets to reduce activity is dipyridamole which also inhibits platelet phosphodiesterases to increase cAMP levels. 130
Yohimbine is an α2-adrenoceptor antagonist and has been shown to be effective at preventing tilt-table test induced syncope, however further testing in this regard is required. 116 While yohimbine in this study was implemented to increase sympathetically driven increases in muscle tone, it may have been effectively preventing the pro-aggregatory stimulus of noradrenaline and adrenaline via platelet α2-adrenoceptors and the release of cardiac vagal afferent chemoreceptor stimulants. It is also worth noting in this study that the α2-adrenoceptor agonist clonidine reduced tolerance to tilt-table induced syncope in 3/8 control patients and all the VVS patients.
While theoretically the β-blockers should be able to prevent catecholamine induced VVS episodes through inhibiting cardiac rate changes, they would not affect the stimulation of α2-adrenoceptors in platelets and may in fact potentiate their pro-aggregatory effects, by non-selective inhibition of the anti-aggregatory β2-adrenoceptors.
SSRIs have been shown to be effective in reducing episodes of the VVS in approximately 50% of patients across the limited trials. The use of the SSRIs for VVS treatment previously described in this report was proposed to alter central levels of serotonin in the NTS. However, in platelets, the inhibition of SERT decreases serotonin storage in the dense granules 132 and chronic SSRI use lowers platelet serotonin content (by 66%) and reduces platelet aggregation responses to ADP, collagen or adrenaline. 133 Hence the actions of the SSRIs may actually be attenuating episodes of VVS through reducing platelet activation and therefore direct activation of vasovagal afferents by locally released serotonin in the heart.
Translation for Pharmacological Treatments for Further Investigation
The optimal medical therapy for patients with VVS is still being defined, with more than one mechanism of various significance among patients.
122
In this study we have reviewed the physiology behind the vasovagal reflex that causes VVS, with respect to neurotransmitters and receptors involved and the source of some of these ligands, see Figure 2. Multiple ligands may also be required to activate the sensory chemoreceptors, future studies into the pharmacological treatment of VVS could focus on preventing the direct activation of cardiac chemoreceptors associated with the vasovagal reflex by stabilizing platelets or inhibiting the chemoreceptor subtypes as follows:
The use of serotonin 5-HT3 receptor antagonists (or SSRIs for comorbidities of depression or anxiety) The use of P2X2/2 receptor antagonists (pain for comorbidities) The use of platelet stabilizers including clopidogrel (inhibit purine P2Y1 receptors), dipyridamole (increased cAMP levels), cilostazol (PDE3inhibitor) or mirabegron (β3-adrenoceptor agonists). May even consider diltiazem or verapamil for cardiovascular disease comorbidities.
The tolerability of serotonin 5HT3 receptor antagonists are considered high with mild constipation and headache as a class adverse effect in several patients. Also QTc elongation has been observed in the number of patients when these medications are applied intravenously. 134 As purinergic P2X3 and P2X2/3 receptors are located on nociceptive neurons, they have potential theoretical uses in pathological conditions such as bladder disorders, gastrointestinal and chronic obstructive pulmonary diseases. An antagonist of these receptors, gefapixant is currently undergoing phase II/III clinical trials for treatment of refractory chronic cough. 135
Conclusion
Catecholamines released in response to stressors, activate a neural reflex process that results in episodes of VVS that may involve platelets as an endogenous source of stimulatory ligands. This review investigates many potential sources of activating neurotransmitters as it may be feasible to inhibit the reflex pharmacologically at several points prior to activation of the cardiac vasovagal afferents through targeting adrenoceptors, serotonin and purine receptors independently or in combination therapies and targeted according to the primary chemoreceptors involved and choices influenced by comorbidities in the patient.
Footnotes
Authors Contribution
Dalyia Abu-Ghazaleh did writing the original draft and revisions. David Taylor did writing—review, editing, and intellectual content. Louise Roberts and Indu Singh did review, editing, and intellectual content. Vinicius Cruzat did writing—review and editing. Roselyn Rose’Meyer did writing the original draft, revisions, and supervision.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability
No data was used for the research described in the article.