
Abstract
Background:
Pulmonary hypertension (PHT) is common in β-thalassemia patients due to hemolysis, iron overload and diminished nitric oxide (NO) levels. Biochemical markers can help to understand the pathophysiology and to introduce new therapies for this condition.
Aim:
This study aimed to evaluate the effectiveness of L-arginine and sildenafil in thalassemia children with PHT at both clinical and biochemical levels.
Methods and Results:
In a randomized controlled study, 60 β-thalassemia major children with PHT were divided into 3 equal groups; Control group (Conventional thalassemia and PHT management), L-arginine group (Conventional + Oral L-arginine 0.1 mg.kg−1 daily), and sildenafil group (Conventional + Oral sildenafil 0.25 mg.kg−1 two times a day) for 60 days. Tricuspid Regurgitant Jet Velocity (TRJV) with Doppler echocardiography along with serum levels of NO, asymmetric dimethylarginine (ADMA), interleukin 1-beta (IL-1β), E-selectin, and visfatin were followed-up at baseline, 30, and 60 days after treatment. Both drugs reduced the TRJV significantly. NO was significantly higher in both L-arginine and sildenafil groups after 60 days compared to baseline, while visfatin levels were lower. Only L-arginine reduced ADMA levels compared to baseline, while sildenafil did not. E-selectin and IL-1β levels did not change remarkably by both drugs. NO and TRJV showed significant negative correlations in both treatment groups.
Conclusion:
L-arginine and sildenafil could clinically ameliorate chronic PHT whereas, L-arginine showed superiority to sildenafil on some biochemical markers.
Keywords
Introduction
Beta-thalassemia (β-thalassemia) syndromes are hereditary blood disorders characterized by defected β globin component of hemoglobin and red blood cells (RBC) synthesis and anemia. It is classified according to the severity into Major, Intermedia and Minor. 1,2
Hemoglobinopathies such as thalassemia and sickle cell disease (SCD) are frequently associated with pulmonary hypertension (PHT). The latter is a serious medical condition in thalassemia patients associated with poor prognosis and increased mortality rate. 3 A Doppler echocardiogram (a non-invasive ultrasound test on the heart) is usually used to identify patients who are at risk for developing PHT. PHT is defined by mean pulmonary artery pressure (MPAP) of more than or equal to 25 mmHg for more than 2 months without association with acute chest syndrome 4 or as a Tricuspid Regurgitant Jet Velocity (TRJV) on Doppler echocardiography >2.5 m/s. 5
Hemoglobin molecules, from destructed erythrocytes, scavenges endothelial nitric oxide (NO), and consequently a state of vasoconstriction, and portal hypertension occur secondary to diminished NO. 6,7 NO is mostly exhausted in the formation of harmful peroxynitrite; one of the reactive oxygen species (ROS) formed from the released free iron after erythrocytes destruction. ROS result also in accumulation of asymmetric dimethylarginine (ADMA) which competitively inhibits NO synthase (NOS). Moreover, NOS itself may produce superoxide instead of NO when arginine concentration becomes low. 6,8 Chronic depletion of NO and arginine in hemolytic anemia, splenectomy, and sticky platelets are common risk factors for portal hypertension. 1 Splenectomy leads to the formation and the aggregation of abnormal erythrocytes, hypercoagulability, and increased adhesion molecules’ expression, which ultimately lead to clinically apparent PHT. 3
Heme, Hb redox forms, and ROS resulting from chronic hemolysis are also associated with vascular endothelial injury that occurs, in most cases, via activation of E-selectin and inflammatory reactions. 9 Consequently, this leads to pro-atherosclerosis and cardiovascular diseases such as PHT. 10 Most of these inflammatory vascular injury markers have been reported to increase in β thalassemia patients. 11,12 Therefore, inflammatory markers such as IL-1β as well as vascular endothelial markers such as serum visfatin and E-selectin levels can be useful as indicators to fully understand this mechanism and to assess the efficacy of potential therapies.
L-arginine is classified as a semi-essential basic amino acid formed from citrulline and ornithine. It is a precursor for nitric oxide by the NOS enzyme. 13 Therefore, the dietary intake of L-arginine (available in red meat, chicken, fish, milk products, and nuts) may be valuable in the body’s response to inflammation and oxidative stress, 8 and reversing endothelial dysfunction. 14 Nutritional supplements containing high L-arginine doses (from 30 to 60 g/day) are still well tolerated. 8 Intravenous and oral L-arginine have been investigated in adult patients with SCD, pulmonary, cardiovascular, and kidney diseases and were reported to be safe. 15
Sildenafil as a potent, orally available phosphodiesterase-5 (PDE-5) inhibitor is also used to treat PHT in the non-thalassaemic population via increasing vasodilatory cGMP. It is one of the FDA-approved medications to treat PHT in adults. 16 The pulmonary vasodilatory effect is observed with both sildenafil monotherapy and when combined with inhaled prostacyclin. 17
Management of PHT in thalassemia patients includes variety of options in adults but not in children. In addition to improvements of transfusion plans, supplemental oxygen, sildenafil can be used as a first-line therapy in adult patients along with warfarin for persistent PHT cases. 18 Standard management of PHT in children with thalassemia relies currently on supportive management such as oxygen therapy to avoid hypoxia, and proper immunization with pneumococcal and influenza vaccines. 19 Clinical evidence for the usefulness of PDE-5, endothelin receptor antagonists, or prostacyclin analogues in those children is sparse and lacks comprehensive data. 3 Medium and high doses of sildenafil showed efficacy in clinical trials performed on children with non-thalassemic PHT conditions. 20 However, unlike other PHT patients, thalassemia patients can have serious complications that may affect the way they respond to certain medications. Due to the complexity of thalassemia pathophysiology, multiple organs can be affected simultaneously with many comorbidities. 21 This emphasizes the need for not only an effective medication but also a drug with high level of safety. Therefore, studies offering therapeutic options to treat PHT in children with thalassemia are urgently needed.
L-arginine and sildenafil are mechanistically different vasodilators which have shown efficacy for similar conditions but do not currently have label indications for use in thalassaemic children. Accordingly, the primary hypothesis of this study is that the off-label monotherapy with either L-arginine or sildenafil for 2 months can improve PHT condition in pediatric patients with a β-thalassemia major assessed by the ability to lower TRJV levels. The secondary hypothesis is that these drugs will counteract the disease pathology by improving the serum levels of some biochemical markers for hemolysis (NO metabolites and ADMA), inflammation (IL-1β), and endothelial dysfunction (Visfatin and E-selectin).
Methods
Study Design
This study is a randomized, prospective, comparative, parallel, controlled clinical trial. One hundred six patients were enrolled at the beginning of the study, inclusion and exclusion criteria were applied. Thirty patients either did not match the criteria or declined to participate. Sixteen patients were lost during the follow ups due to changing the transfusion center, skipping sampling time, or due to the poor adherence. Sixty age, sex, body mass index, and ethnicity matched children with β-thalassemia major continued the study. The CONSORT flow chart is provided in Figure 1.

CONSORT flow chart showing the enrollment, randomization, and follow-up of patients through the trial as per the criteria reported in the CONSORT Guidelines.
The study was registered in www.clinicaltrials.gov (NCT03402191), approved by Tanta University ethical committee (approval code: 31766/09/17), and was conducted based on the guidelines of the Declaration of Helsinki. Written informed consent was signed by the guardian of the children. All participants were recruited from the Hematology Unit of the Pediatric Department, Tanta University Hospital.
Inclusion criteria were children with β-thalassemia major and having increased TRJV higher than 2.5 m/s. Both sexes with an age range of 6-18 years old. Children diagnosed with a stable stage of their disease at the evaluation time were enrolled in the study. All patients in the study received regular transfusions every 2-4 weeks. The exclusion criteria were other chronic hemolytic anemia, young age (<6 years old), hypotension (BP less than the 5th percentile for age), other heart diseases (either congenital or rheumatic), inability to take or tolerate oral medications, allergy to arginine or sildenafil, patients on or were previously treated with medications that can reduce pulmonary hypertension (e.g., PDE-5 inhibitors, endothelin receptor antagonists, or prostaglandin agonists), diabetic patients, pregnancy, patients who received a blood transfusion on the same day prior to the collection of samples, patients who received hydroxyurea within the 90 days prior to the study entry, patients with pulmonary stenosis or other structural occlusion affecting pulmonary blood circulation or flow, signs of left heart failure (e.g., fraction shortening less than 28% and ejection fraction lower than 50%), mitral valve regurgitation (grades 2 to 4), or mitral valve stenosis, arrhythmias (e.g., atrial fibrillation or ventricular tachycardia), and severe pericardial effusion.
Seventy-six patients who met the inclusion criteria and provided the written consent forms were randomized by stratified random block method into 3 groups; Group I, or the control group (n = 25), included children with β-thalassemia and PHT who received neither L-arginine nor sildenafil therapy, Group II (n = 26): which involved children with β-thalassemia major associated with PHT who received oral L-arginine with a daily dose of 0.1 mg.kg−1 for consecutive 60 days, and Group III (n = 25) that included children with β-thalassemia major associated with PHT who received oral sildenafil therapy in a dose of 0.25 mg.kg−1 two times daily for consecutive 60 days. All patients were followed-up regularly and the drugs were provided on week-by-week bases to monitor the prescription refill rate and ensure good adherence to therapy protocol.
Patient History and Demographics
All children in the 3 study groups were subjected to: Demography and anthropometric measurements including age, sex, measurement of weight and height using Detecto scale and calculation of body mass index; BMI = Weigh (kg)/height
2
(m). History taking from patient records including duration of anemia and its related symptoms and complications. Complete clinical examination including inspection of the overall appearance (including skin color and condition, as well as the extremities), abdominal examination (such as signs of hepato-splenomegaly and enlargement of other organs), and systemic examination (including chest, heart, CNS, and genitourinary system).
Biochemical Tests
Laboratory analysis was performed on venous blood collected from anticubetal vein at morning between 9-10 AM to avoid diurnal variation. These blood samples were collected at baseline (before initiation of any treatment) and then 30 and 60 days after treatment with the medications under study. Only 60 patients out on the randomized 76 patients (20 patients in each study arm) continued the follow-up to the end of the 60 days of treatment due to 5-6 patients drop-out from each group (Figure 1). Samples were collected from each patient at the predefined time points for the assessment of the following parameters: Assessment of routine parameters including complete blood count, hemoglobin concentration, serum ferritin level, liver function (ALT, AST, total and direct bilirubin), and kidney function (serum creatinine). These routine parameters were assessed at baseline using standard methodologies. Assessment of follow-up parameters Parameters related to oxidative stress as a negative regulator for erythropoiesis including nitric oxide (NO) which were assessed calorimetrically using the method described by Montgomery and Dymock method
22
and by a commercial kit (Biodiagnostic Company, Giza, Egypt). Serum asymmetric dimethyl arginine (ADMA) level which was assessed using commercially available kit in accordance with the manufacturer’s instructions (SunRed Biotechnology Company, Shanghai, China; Cat #. 201-12-1888). Parameters related to endothelial dysfunction as a complication for thalassemia including serum E-Selectin and visfatin which were measured using commercially available ELISA kits in accordance with the manufacturer’s instructions (SunRed Biotechnology Company, Shanghai, China; Cat #. 201-12-0254, and 201-12-0026 respectively). Parameter related to inflammation as a negative regulator for erythropoiesis including human IL-1β which was assessed in plasma samples using double-antibody sandwich ELISA kit in accordance with the manufacturer’s instructions (SunRed Biotechnology Company Shanghai, China; Cat #. 201-12-0144).
Echocardiography
Echocardiography was carried-out at Tanta University Hospital, Paediatric Department, Paediatric Echocardiography Laboratory using Ultrasound Machine (Vivid 7, G.E.,) in accordance with a standardized protocol following the guidelines of the American Society of Echocardiography. 23
Children who were previously diagnosed with PHT were included directly in the study. A TRJV cut-off level of 2.5 m/s was considered for the diagnosis of PHT.
TRJV was assessed by measuring peak systolic right ventricular and right atrial pressure gradient using both pulsed- and continuous-wave Doppler echocardiography tracing. The optimal tricuspid Doppler flow signal was obtained after recording different apical 4-chamber views with at least 5 sequential signals. Normally, the trans-tricuspid gradient and the right atrial pressure are summed to calculate the pulmonary artery systolic pressure (PASP). Children included in the present study did not have heart failure and the mean right atrial pressure was below 5 mmHg, therefore it was ignored. Accordingly, PASP was nearly equal to the tricuspid regurgitation in those compensated patients. 24
Echocardiograms were performed for the 3 groups before initiation of study (day 0) and then 30 and 60 days after the commission of the study and the initiation of the study medications in the treatment groups.
Statistics
All data are represented as mean ± SD. Comparison between the 3 groups was performed using Kolmogorov-Smirnov test was used to test the normality of data. Data was considered normally distributed when P value ≥.05. Chi-Square test was used for statistical analysis of nominal data. One-way ANOVA was used for parametric statistics. P value <.05 was considered statistically significant.
For non-parametric statistics, median values [95% confidence interval; CI] were reported and Kruskal-Wallis test was used to compare control, L-arginine, and sildenafil groups. Within each treatment group, Friedman test was used to compare the changes within the same treatment group at the baseline, after 30 days, and after 60 days of treatment. Pairwise multiple comparison was performed using Dunn’s test. The level of significance was set at P < .05.
Correlation between variables was evaluated by Spearman correlations. Correlations were considered weak when Spearman correlation coefficient (rs) was 0.2-0.39, moderate for rs = 0.4-0.59, and strong rs ≥ 0.6. The statistical analysis was performed using GraphPad Prism v9. The level of significance was set at P < .05.
Results
Patient Demographic and Anthropometric Data
No statistically significant differences were observed among the 3 study groups with regard to age, body weight, BMI (P > .05), and sex ratio (Chi-square < df) as shown in Table 1.
Demographic and Anthropometric Data of the 3 Study Groups.
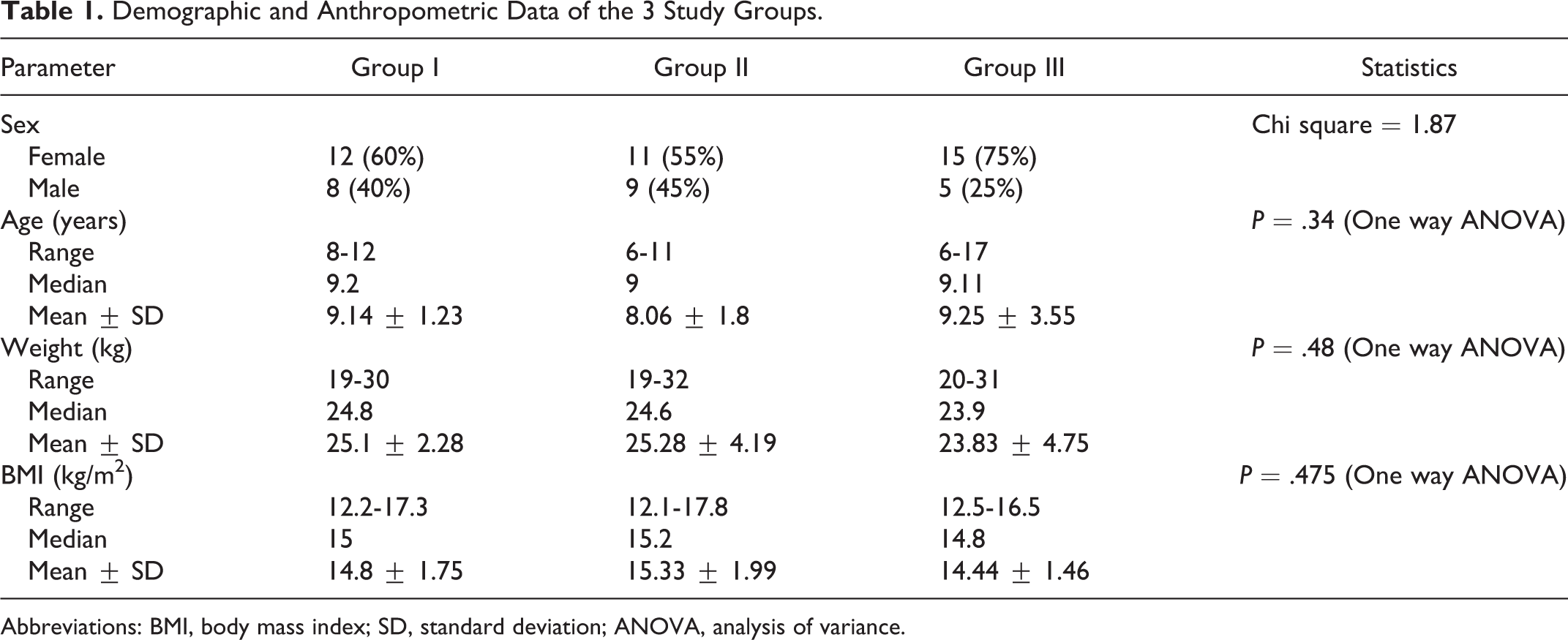
Abbreviations: BMI, body mass index; SD, standard deviation; ANOVA, analysis of variance.
Routinely Measured Parameters
Routine parameters for thalassemia also showed non-significant variations among patients of the 3 study groups at the beginning of the study (Table 2).
Routinely Measured Biochemical Parameters at Baseline.a

Abbreviations: MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; RBCs, red blood cells.
a Samples for these biochemical tests are collected pre-transfusion.
Results of Follow-Up Parameters
Nitric oxide assay results
Kolmogorov-Smirnov normality test showed P < .05 for some groups. Therefore, non-parametric statistics were used. At the baseline, there were non-significant differences in nitric oxide (NO) metabolite levels among the 3 treatment groups. Both L-Arginine and sildenafil showed a gradual rise in NO levels after 1 and 2 months of treatments (Figure S1). Two months of treatments resulted in a significant increase in the median NO levels by ∼

Results obtained with biochemical markers (A: Nitric oxide, B: ADMA, C: Visfatin, D: E-selectin, and E: IL-1β) comparing Control, L-Arginine, and Sildenafil groups at baseline and 2 months after treatments. Red stars represent significant difference (P < .05) compared to the corresponding baseline levels for the same group. Black arrows represent the direction of these changes.
ADMA assay results
At baseline, there were non-significant variations in ADMA levels among all study groups. Compared to baseline data, the control group showed gradual and significant rises in ADMA levels after 1 month (by
Visfatin assay results
At baseline, there were non-significant variations in visfatin levels among all study groups. Compared to baseline data, the control group showed gradual and significant rises in visfatin levels after 1 month (by
E-selectin assay results
There were non-significant differences in E-selectin levels among the 3 study groups at any time of treatment. For the same patients, no significant change was noticed in any study group over the time-course of treatments. Moreover, the study groups showed high inter-individual variabilities in E-selectin levels (Figure 2D).
IL-1β assay results
At the baseline, there were non-significant variations among the 3 study groups regarding IL-1β levels. There was a gradual increase in IL-1β level for the control group over the time course of the study that reached up to
Doppler echocardiography results
No statistically significant differences were noticed in TRJV among the 3 groups at the baseline. No significant differences were observed between L-arginine and sildenafil treated groups at all-time points of the follow-up period (P > .05). For the control group, the statistical analysis using Friedman test followed by Dunn’s test demonstrated that there was a gradual and significant increase in TRJV median values [95% CI] after 1 (4.3 [3.4-5]) and 2 months (4.6 [3.7-5]) of the follow-up period (up to

Tricuspid Regurgitant Jet Velocity in m/s determined by Doppler echocardiography for Control, L-Arginine, and Sildenafil groups at baseline and after 2 months of treatments. Red stars represent significant difference (P < .05) compared to the corresponding baseline values for the same group. Black arrows represent the direction of these changes.
Correlations Among Follow-Up Parameters
Correlation analyses among all monitored parameters revealed the presence of strong negative and significant correlations between serum NO metabolite levels and TRJV in both L-arginine and sildenafil treated groups. Positive significant correlation was observed between E-selectin and visfatin (Table 3).
Spearman’s Correlation Among the Follow-Up Parameters in Both L-Arginine and Sildenafil Treated Groups.a

a Shaded area refers to correlations for sildenafil group while the clear area represents correlations for L-Arginine group. Cells with thick borders and stars are those with significant correlations, and × in the diagonal line refers to the unity when the marker was correlated to itself.
Discussion
Many patients with thalassemia suffer from PHT which affects about 10% to 75% in patients with thalassemia major. 3,25 The main causes of PHT are related to hemolysis, iron overload, and splenectomy. Biochemically, PHT is linked to arginine dysregulation and reduced NO level. 26 Exploring effective vasodilators for the management of this condition in such lifelong disease is an urgent necessity and will open an avenue for novel therapeutic strategies especially in pediatric population. Based on the pathogenesis of the condition, L-arginine and sildenafil were proposed as promising medications for the management of this increase in pulmonary arterial pressure. Although sildenafil is an approved therapy by FDA for the management of pulmonary hypertension in general, 27 it has not yet been approved for children with thalassemia.
The current study revealed the usefulness of the 2 medications both clinically and biochemically. Patients included in this study have relatively lower pre-perfusion hemoglobin concentration levels compared to other studies that can be attributed to ethnicity, epidemiology, and the lower rate of blood donation as this blood is normally given to patients for free. 28 A previous study by our group showed the effectiveness of both L-arginine and sildenafil in reducing significantly the TRJV after 1 month of therapy. 29 One limitation of our previous study was the short duration of treatment with both drugs which was only 1 month. In the current study the duration of treatment was doubled to 2 months with 3 follow-up time points (Baseline, after 1 month, and after 2 months) to unveil possible differences in the effectiveness of the 2 medications. Moreover, the current study provides a mechanistic explanation of such effect or differences with the biochemical assays of the biomarkers involved in the pathogenesis of PTH.
Various TRJV cut-off points were adopted in many studies (2.5 m/s, 2.9 m/s, or 3.0 m/s) which resulted in variability in the disease frequencies. During the current study and at baseline, all patients in the 3 study groups had TRJV values greater than 3.0 m/s which reduce the likelihood of misdiagnosis of pulmonary hypertension. 30 Both medications produced a gradual clinical effectiveness by reducing TRJV after 1 and 2 months of therapy to be less than 3 m/s in both groups by the end of the study. This result is in accordance with some previous studies demonstrated the efficacy and safety of both drugs in adult patients with hemoglobinopathies. 26,31,32 The same dose of L-arginine was previously administered, for 5 days, to 10 SCD patients with PHT. 6 It resulted in lowering the mean pulmonary artery pressures by about 15% concomitantly with an improvement of venous oxygen saturation. This former study suggests that arginine could affect pulmonary as well as systemic vascular resistance. 6 In our study, a higher reduction in TRJV values was observed in thalassaemic patients (up to 35%) which might be attributed to the longer period of treatment.
L-arginine showed superiority over sildenafil regarding its effect on ADMA serum levels. These data confirm the mechanism through which L-arginine improved PHT condition through reducing the serum level of ADMA and consequently counteracting its inhibitory effect on NOS. This outcome is supposed to be useful for many chronic disease conditions 33 including thalassemia as proved herein. Although both L-arginine and sildenafil did not affect the level of IL-1β (an inflammatory marker) compared to their baseline values, they silenced the gradual propagation of its levels that was noticed with the control group. Therefore, they might be useful in preventing thalassemia associated-inflammation which is accused for enhancing apoptosis and shortening the lifespan of circulating red blood cells in thalassaemic patients. 34
Sildenafil increased gradually NO metabolite levels over time which agrees with the molecular biology and micromorphological studies that showed the ability of sildenafil to inhibit the breakdown of cyclic guanosine monophosphate by inhibiting phosphodiesterase-5 leading to an increase in the intracellular NO levels with some impact on the extracellular secretion of NO into blood. 35,36
Neither L-arginine nor sildenafil provoked any change in both E-selectin levels throughout the treatment duration. Normal range of serum E-selectin levels in healthy children as documented in scientific literature is 36.7-153.2 ng/mL with an average value of 79.7 ng/mL. 37 In the current study, the baseline level of serum E-selectin in patients with β-thalassemia major was 25.2-449.6 ng/mL with an average value of 153.4 ng/mL. This average value is nearly 92% higher than its average value reported in healthy children indicating endothelial cells activation. Both sildenafil and L-arginine failed to reduce this endothelial activation or dysfunction despite the 2 months treatment period. In this context, further longitudinal studies are still required to investigate the impact of longer-term treatment of both L-arginine and sildenafil on adhesion molecules serum levels.
The findings obtained with the current study showed higher mean serum visfatin level for the recruited thalassemic patients (average value of 7.1 ng/mL and a range of 1.9-13.4 ng/mL) than that reported in the literature for normal weight healthy children (average value of 2.19 ng/mL and a range of 1.4-3.26 ng/mL). 38 The current results come in line with previous studies postulated higher level of adipocytokines notably visfatin among patients with β-thalassemia. It also indicates the possible inflammatory role of visfatin in this condition, the possible association of this adipocytokine with endothelial and vascular dysfunction in patients with β-thalassemia, and its correlation with disease severity. 11,12 Both L-arginine and sildenafil reduced visfatin serum level after 2 months of treatment. This effect can be partly attributed to the direct anti-inflammatory effect or the ability of these drugs to prevent the progression of inflammation through vaso-modulatory mechanisms similar to that reported previously in other chronic inflammatory diseases. 39
As control group was only receiving the standard care, levels of ADMA, IL-1β and visfatin as well as TRJV went up after 2 months from starting the study. This means that regular transfusion can help, but according to current study results, it might not be enough and requires additional therapeutic options. Although the change in the control group was higher than expected over this short period of time, previous studies including a placebo group has pointed out to the progressive nature of PHT especially in thalassemia patients. 40 An average increase in MPAP was reported to be 5%-8% over 12 weeks in adult thalassemia patients, however most of those patients were already on sildenafil therapy. 41 Our study showed an average change in TRJV of 15% over 8 weeks only which might be attributed to the small sample size, young age, variable time since the onset of the disease, and splenectomy status along with the progressive nature of the disease with standard interventions. Moreover, the change in TRJV was inversely proportional to baseline levels which indicate that this change is not linear (not steadily increasing; Figure S2), and some compromising mechanisms with the regular transfusion might be taking place at high TRJV levels. However, patients are still in need for specific PHT treatments to avoid long term cardiovascular complications.
As sildenafil is known to be reducing NO independent on ADMA mechanism, the median ADMA levels seem to be increasing after 2 months similar to the control group. However, this was not reflected statistically due to the high variability in sildenafil groups’ ADMA levels and the relatively low sample size.
The negative significant correlations between NO metabolite serum level and TRJV support the theory and the mechanism related to improving pulmonary arteries constriction (Lowering TRJV) via increasing the levels of NO which was drastically reduced due to hemolysis. Both L-arginine and sildenafil treated groups showed positive significant correlation between E-selectin and visfatin levels. These results seem to be in parallel with some previous studies demonstrating the ability of visfatin to induce inflammatory response in endothelial cells which is associated with high levels of various soluble adhesion molecules. 42,43
It is worth mentioning that the novelty of the current study includes the association between clinical assessments with the biochemical parameters which were not routinely measured in children with β thalassemia major after being treated with the study medications. Furthermore, the points of strength of the current study include its randomized and comparative design and the assessment of patients at 3 time points. However, the study has some limitations including the relatively small sample size which was difficult to overcome in such rare disease.
Conclusion
Both L-arginine and sildenafil were well-tolerated with the implicated doses for children with β-thalassemia major. Additionally, both medications showed clinical efficacy on pulmonary arterial pressure which was assessed by Doppler echocardiography. L-arginine showed better improvements in some of the assessed biochemical parameters compared to sildenafil especially those related to ADMA levels which can explain its unique mechanism in regenerating endothelial nitric oxide. Despite our promising results, large scale and more longitudinal studies are still needed along with pharmacoeconomic studies to account for benefit/cost ratio that might differ from country to another based on the availability of both medications.
Supplemental Material
Supplemental Material, sj-docx-1-cpt-10.1177_10742484221132671 - Randomized Clinical and Biochemical Study Comparing the Effect of L-arginine and Sildenafil in Beta Thalassemia Major Children With High Tricuspid Regurgitant Jet Velocity
Supplemental Material, sj-docx-1-cpt-10.1177_10742484221132671 for Randomized Clinical and Biochemical Study Comparing the Effect of L-arginine and Sildenafil in Beta Thalassemia Major Children With High Tricuspid Regurgitant Jet Velocity by Eman El-Khateeb, Sahar Mohamed El-Haggar, Osama El-Razaky, Mohamed Ramadan El-Shanshory and Tarek Mohamed Mostafa in Journal of Cardiovascular Pharmacology and Therapeutics
Footnotes
Authors’ Note
Acknowledgments
Special thanks to the patients and their carers who agreed to participate in this study. Authors also acknowledge the nursing staff and the healthcare team in the pediatric unit at Tanta University Hospital for facilitating the use of instruments and the environment for this research and for their valuable help and recommendations. Authors would like to acknowledge EIPICO pharmaceutical company for providing us with half the sildenafil quantity required for this research.
Author Contributions
All authors reviewed the literature and constructed the study design. Eligibility and enrollment of the study participants was done by Eman El-Khateeb, Osama El-Razaky and Mohamed Ramadan El-Shanshory. Laboratory analysis was done by Eman El-Khateeb and Tarek Mohamed Mostafa. Doppler echocardiography was performed by Osama El-Razaky and Eman El-Khateeb. Statistical analysis was done by Eman El-Khateeb and Sahar Mohamed El-Haggar. All authors wrote, revised, and approved the final version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was sponsored by the Egyptian government (research development funds to universities and research sectors) in addition to the self-funding by the corresponding author.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.