
Abstract
Vascular calcification results from an imbalance of promoters and inhibitors of mineralization in the vascular wall, culminating in the creation of an organized extracellular matrix deposition. It is characterized by the accumulation of calcium phosphate complex and crystallization of hydroxyapatite in the tunica media, leading to vessel stiffening. The underlying initiators of dysregulated calcification maintenance are diverse. These range from the expression of bone-associated proteins, to the osteogenic transdifferentiation of smooth muscle cells to osteoblast-like cells, to the release of fragmented apoptotic bodies and mineralization competent extracellular vesicles by smooth muscle cells, which act as a nucleation site for the deposition of hydroxyapatite crystals. The process involves a complex interplay between vitamin K-dependent calcification-inhibitory proteins, such as matrix γ-carboxyglutamate acid (Gla) protein, Gla-rich protein and growth arrest-specific gene 6 protein, and stimulatory mediators, such as osteocalcin. Vitamin K plays an important role as a cofactor for posttranslational γ-carboxylation of matrix Gla proteins in converting to a biologically active conformation. Drugs that inhibit vitamin K, such as warfarin, impair γ-carboxylation of Gla proteins, resulting in the accumulation of uncarboxylated proteins lacking calcification-inhibitory capacity. This article overviews the involvement of systemically and locally expressed vitamin K-dependent proteins in vascular calcification and their potential as biomarkers of calcification.
Keywords
Introduction
Vascular calcification develops from a disruption of an actively regulated process that induces ossification, which is driven principally by medial smooth muscle cells. 1,2 Vascular smooth muscle cells are of mesenchymal origin and under stress conditions they transdifferentiate to multipotent cell types, such as osteoblasts and chondrocytes. 3 Smooth muscle cells exhibit multilineage plasticity that allows induction of osteogenic differentiation, with subsequent conversion from contractile to synthetic and osteogenic cell phenotypes. These are characterized by loss of smooth muscle cell phenotype and increased expression of osteogenic genes. 4,5
Vascular smooth muscle cell calcification is represented by osteoblast-like cells with deposits of crystallized hydroxyapatite in the extracellular matrix. 2 It involves osteochondrogenic transdifferentiated cells, downregulation of calcification inhibitors, upregulation of bone mineralization-regulatory genes, and release of calcifying membrane-bound vesicles from smooth muscle cells. 4 In addition, the transition into osteoblast-like cells is enhanced by an increase in intracellular phosphate concentration, which occurs in response to hyperphosphatemia. 3 Hyperphosphatemia is observed in chronic kidney disease due to decreased renal phosphate excretion and participates in the stimulation of vascular calcification.
Moreover, smooth muscle cell apoptosis, oxidative stress, increased extracellular matrix remodeling, secretion of matrix metalloproteinases, and endothelial dysfunction contribute to the development and progression of calcification. 6,7 Pro-inflammatory cytokines amplify the local inflammatory response of smooth muscle cell differentiation to osteogenic cells, leading to the enhanced calcium loading and vesicle release that create conditions for calcification. Apoptotic smooth muscle cells generate negatively charged membrane particles. These promote initiation of calcification by serving as an initial site for calcium phosphate crystal precipitation. 5
The extracellular fluid is supersaturated with calcium and phosphate; however, spontaneous precipitation of calcium phosphate crystals on the vascular wall does not occur. This is due to a tightly regulated process mediated by vitamin K-dependent calcification inhibitor proteins, such as matrix carboxyglutamate protein (MGP) and γ-carboxyglutamate acid (Gla)-rich protein (GRP). 8 The secreted active protein inhibitors protect the blood vessel from undergoing calcification. This article summarizes the cellular and molecular factors that mediate mineralization and the potential role of vitamin K-dependent proteins in maintaining vascular wall homeostasis (Figure 1).
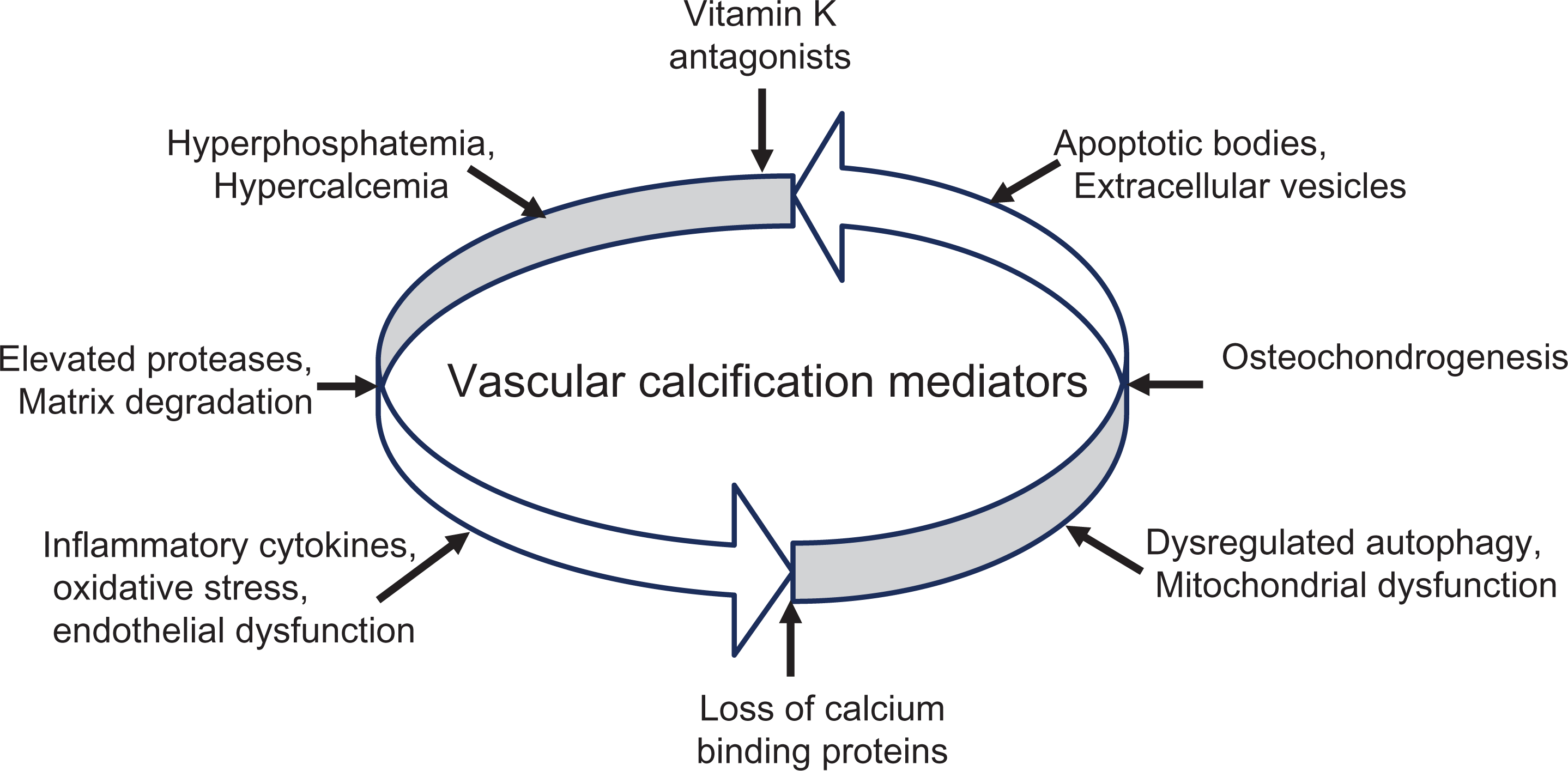
Vascular calcification mediators: Vitamin K antagonists inhibit γ-carboxylation of vitamin K-dependent proteins, leading to vascular calcification. Cell death leads to release of apoptotic bodies that nucleate hydroxyapatite at injury sites. Deficiency of constitutively expressed tissue-derived and circulating mineralization inhibitors leads to hydroxyapatite deposition and induction of bone formation resulting from differentiated smooth muscle cells. Under calcification promoter conditions, such as inflammation, oxidative stress, vitamin K deficiency, or aging, smooth muscle cells downregulate the expression of contractile-associated genes and upregulate the expression of osteogenic markers, acquiring a synthetic phenotype. These events culminate in the osteochondrogenic differentiated smooth muscle cells and the release of calcifying extracellular vesicles, characterized by hydroxyapatite nucleation crystals, cell apoptosis, and reduced calcification inhibitors. Increased bone-associated proteins and the deposition of calcifying-competent membrane vesicles ultimately lead to mineralization.
Subtypes of Vascular Calcification
Vascular calcification can be morphologically classified into two distinct subtypes depending on its location, that is, within the intimal layer or the medial layer (Table 1). Intimal calcification occurs on the surface of atherosclerotic plaques and indicates plaque burden and luminal narrowing, whereas medial calcification is associated with the muscular layer and manifests as stiffness and reduced vascular compliance.
Subtypes of Vascular Calcification.

Intimal Calcification
Intimal or atherosclerotic calcification is initiated by chronic inflammation and lipid accumulation, with dyslipidemia often associated with the extent of calcium deposits. It involves inflammatory mediators, phagocytosis of hydroxyapatite crystals, and expression of cytokines and chemokines. 1 Intimal calcification is manifested as patchy calcification within lipid deposits and is represented as eccentric remodeling of plaque calcification, reduced lumen diameter and thickening of the intimal layer. 9 Intimal atherosclerotic calcification progresses to punctate lesions, accompanied by apoptosis of smooth muscle cells, and release of extracellular vesicles. Intimal calcification is mostly observed in the coronary arteries.
Endothelial cells participate in intimal calcification by a mechanism that involves endothelial-to-mesenchymal transition to osteochondrogenesis. 10,11 In the process of transition, the transformed cells gain multipotency and thereafter differentiate into an osteoblast-like cell lineage. 11 Stimulation of endothelial cells using transforming growth factor-β (TGF-β) leads to phenotypic changes of endothelial-to-mesenchymal-like transition, with subsequent increases in the expression of procalcific mediators, such as osteocalcin and Runt-related transcription factor 2 (Runx2). 12,13 The association of endothelial dysfunction with calcification indicates that endothelial-derived nitric oxide plays a role in limiting vascular calcification by inhibiting signaling mediators of the phenotypic switch to osteoblast-like cells. 14
Medial Calcification
Medial calcification is a type of arteriosclerosis, characterized by a concentric deposition of calcium in the tunica media and its process is distinct from that of intimal atherosclerotic calcification. It occurs along the elastic lamellae between the smooth muscle cells and elastin layers, in the absence of lipid or inflammatory cells. Medial calcification is described in terms of arterial hardening, loss of elasticity, and stiffness. 15 It is more frequently observed in the peripheral arteries and is less abundant in coronary artery disease.
Medial calcification is driven by differentiation of smooth muscle cells into osteoblast-like cells, upregulation of bone-associated proteins such as osteocalcin, and an increase in bone-specific alkaline phosphatase (ALP) activity. 4 It consists of calcium phosphate complexes in the form of hydroxyapatite crystals, primarily contributed by osteochondrogenic differentiation of smooth muscle cells. 3 Osteochondrogenic smooth muscle cells secrete fewer inhibitory Gla proteins and become susceptible to apoptosis, releasing apoptotic bodies and vesicles that promote calcification. Also occurring are matrix remodeling events, including the degradation of extracellular matrix and elastin by matrix metalloproteinases, followed by calcium deposition onto degraded elastin. 3,16
In addition to smooth muscle cells, osteoblast-like cells can also be derived from myofibroblasts in the adventitia induced by TGF-β1. 17 The notion that the adventitia initiates the calcification process is based on previous reports which demonstrated that surgical resection of the periadventitial layer prevents medial artery calcification. 17 Activated adventitial fibroblasts differentiate into migratory myofibroblasts and participate in the process of adaptive fibrotic remodeling consisting of structural and functional reorganization. In addition, adventitial stem and progenitor cells have multilineage plasticity with the potential to differentiate into osteogenic cells.
Cellular Mediators of Vascular Calcification
Smooth muscle cells initiate and regulate calcification by such mechanisms as upregulation of osteogenic proteins, release of calcifying extracellular vesicles, and fragmentation of apoptotic bodies that serve as sites for the accumulation of calcium phosphate complexes.
Smooth Muscle Cell Differentiation to Osteogenic Phenotype
Under pathologic conditions leading to an imbalance between promoters and inhibitors of calcification, smooth muscle cells undergo phenotypic transition characterized by the loss of contractile specific marker genes, such as smooth muscle 22 α and α-smooth muscle actin, and expression of bone-related genes, such as Runx2, bone morphogenetic protein-2 (BMP-2), and osteocalcin. 3 The transcription factor Runx2 plays a major role in regulating osteoblastic and chondrocytic differentiation in calcified vascular lesions. 3,18 Under conditions that promote calcification, smooth muscle cells downregulate the expression of contractile-associated genes and upregulate the expression of osteogenic markers, such as Runx2, and gain a synthetic phenotype that promotes calcification. 18 In addition, other factors create conditions that facilitate the phenotypic switch of smooth muscle cells, such as inflammation, oxidative stress, aging, vitamin K deficiency and paracrine stimuli by TGF-β and fibroblast growth factor). 4
Differentiation of vascular smooth muscle cells into phenotypically distinct osteoblast-like cells is characterized by the development of calcifying vesicles and downregulation of calcification inhibitors. 2 In addition, elevated phosphate levels upregulate Runx2 and promote osteo/chondrogenic transdifferentiation of smooth muscle cells, enhancing the calcification process. 16 Furthermore, transformed smooth muscle cells increase ALP expression, which degrades the calcification inhibitor pyrophosphate and creates a phosphate source, leading to hydroxyapatite formation. 13
Extracellular Vesicles as Mineral Nucleation Sites
Smooth muscle cells differentiated to osteogenic phenotypes release small membrane-encapsulated vesicles containing cellular components enriched with pro-mineralizing factors. 1,7 Extracellular vesicles are formed from direct budding of the plasma membrane or through fusion of endosomal vesicles with plasma membrane, and subsequent expulsion of intraluminal vesicles. 12,19 The calcification-competent extracellular vesicles increase calcium deposition, along with the release of apoptotic bodies serving as nucleating sites for hydroxyapatite and crystal growth. 5,7,20 Apoptotic bodies are formed during smooth muscle cell apoptosis due to cell blebbing with cellular contents released into the extracellular space.
Extracellular vesicles efficiently accumulate calcium and phosphate to form hydroxyapatite crystals. They participate in hyperphosphatemia-induced calcification by a mechanism involving mineral nucleation and osteogenesis. Hypercalcemia and hyperphosphatemia induce apoptosis and enhance formation of mineral nucleation complexes. High cytosolic calcium concentration stimulates the translocation of annexins to the plasma membrane, activating the release of vesicles and converting them to mineralization-competent extracellular vesicles. These have small phospholipidic membrane–bound particles with the capacity to nucleate calcium and phosphate. 2 Reuptake of calcified vesicles by smooth muscle cells enhances apoptosis, leading to further calcification.
Under normal conditions, smooth muscle cell–derived membrane vesicles do not calcify because they contain mineralization inhibitors. Extracellular vesicles contain reduced amounts of calcification inhibitor proteins and exhibit unrestricted calcium loading capacity. In the absence of active calcification inhibitors, mineralization-competent extracellular vesicles initiate calcification by efficiently nucleating hydroxyapatite formation in the media. 2,7 Extracellular vesicles and remnant apoptotic bodies released by smooth muscle cells act as mineral nucleation sites for the deposition of calcium and phosphate in the vascular wall.
Adaptive Response
To promote survival and react to stress, vascular smooth muscle cells undergo autophagy through a regulated process of self-digestion and removal of unnecessary cellular components. By activating autophagy, smooth muscle cells overcome toxic insults by allowing the removal of toxic products, promoting their survival. An autophagic response prevents the release of apoptotic bodies and extracellular vesicles and functions as an endogenous protective mechanism to attenuate calcification and maintain cellular homeostasis. 20
Autophagy protects against phosphate-induced smooth muscle cell calcification by regulating apoptosis and the release of calcifiable membrane vesicles. 20 It also regulates the process of smooth muscle cell plasticity and phenotypic changes. Dysregulated autophagy enhances the development of stress-induced senescence and calcium mobilization. 2,15 In addition, mitochondrial autophagic dysfunction contributes to the development of vascular calcification through an intrinsic apoptosis pathway, which activates caspase-9 and enhances senescence by generating reactive oxygen species.
Induction of autophagy in smooth muscle cells protects against cell death, stimulates cell survival, and inhibits the conversion of contractile smooth muscle to a synthetic function. 20,21 Activation of autophagy protects medial smooth muscle cells by inhibiting differentiation into osteogenic phenotypes. 21 Drugs that activate autophagy, such as atorvastatin and telmisartan, protect TGF-β stimulated calcification of smooth muscle cells, suggesting that induction of autophagy might have a therapeutic application to reduce vascular calcification. 20 –22 Downregulation of autophagy enhances the release of phosphate-induced procalcific vesicles and increases ALP activity. 20 Agents that block autophagy, such as 3-methyladenine and bafilomycin, increase calcification by enhancing apoptotic response, and agents that induce autophagy protect cells and mice from uremic media calcification by inhibiting osteogenic transdifferentiation of smooth muscle cells. 20,21 Impaired autophagy initiates the development of stress-induced premature smooth muscle cell senescence, as well as the loss of contractile proteins and phenotypic changes that promote calcification. 15 In short, induction of autophagy plays a cytoprotective role of suppressing smooth muscle cell senescence and preventing calcification, whereas inhibition of autophagy enhances the release of apoptotic bodies and promotes calcification.
Calcium-Sensing Receptors
The calcium-sensing receptor is expressed in the vasculature and functions as a sensor for changes in extracellular calcium concentration. Exposure of smooth muscle cells to high-extracellular calcium concentration downregulates the expression of calcium-sensing receptors. 23 Activation of calcium-sensing receptors inhibits induction of osteochondrogenic transformation of smooth muscle cell phenotypes. 23,24 In addition, activation of calcium-sensing receptors stimulates matrix-Gla protein and downregulates osteoblast-specific gene BMP-2, promoting anticalcific effects. 25,26 Upregulated expression of a functional calcium-sensing receptor inhibits transformation toward a calcifying smooth muscle cell phenotype, protecting against calcification. Drugs that enhance the sensitivity of the calcium-sensing receptor to calcium decrease calcification, and exert vasculoprotective effects. 23,27 Calcimimetics, which allosterically activate the calcium-sensing receptor, exert antiapoptotic effects and enhance the proliferative potential of smooth muscle cells under stress conditions. Calcimimetic compounds reduce serum parathyroid hormone, calcium, and phosphate in dialysis patients. 28
Ablation of calcium-sensing receptor function reduces the proliferative potential of smooth muscle cells and increases calcification in response to calcium and phosphate. 27,28 Downregulated expression of the calcium-sensing receptor enhances differentiation of smooth muscle cells into osteogenic phenotypes, with a concomitant increase in calcification. 23 Calcium-sensing receptors regulate parathyroid hormone synthesis, and its downregulation is associated with abnormal parathyroid hormone secretion and hypercalcemia in chronic kidney disease. 29 Thus, the expression of calcium-sensing receptors in the vasculature is inversely associated with calcification.
Vitamin K Cycle in Vascular Calcification
The vitamin K cycle plays an essential role in the regulation of proteins associated with vascular calcification. Vitamin K acts as a coenzyme for the enzyme γ-glutamyl carboxylase, which catalyzes glutamic acid (Glu) residues into Gla. 9 It is required for posttranslational modification of protein-bound Glu’s conversion to the γ-carboxyglutamate active conformation form, which functions as an anticalcific or procalcific. 30 The γ-carboxylation reaction converts a reduced form of vitamin K hydroquinone to the epoxide form of vitamin K and forms an active Gla residue.
Vitamin K exists in 2 biologically active forms that include vitamin K1 (phylloquinone) and vitamin K2 (menaquinone). Vitamin K1 is primarily found in green vegetables or vegetable oils, and vitamin K2 is most abundant in fermented foods, meat, egg yolk, and dairy products. Vitamin K1 has higher effect in preventing excessive bleeding, and vitamin K2 has higher effect in reducing vascular calcification because of its prolonged activity in the body. 31 To maintain a suitable vitamin K balance, daily dietary intake of 90 µg/d for female and 120 µg/d for male are recommended. These levels may be adequate to enable proper blood clotting but fall below the levels needed to protect against vascular calcification. 32
The vitamin K-dependent carboxylase is an integral membrane glycoprotein that uses vitamin K to modify glutamyl residues to γ-carboxylated glutamyl residues, a metabolically active form. The carboxylation is coupled to oxidation of vitamin K during the addition of the γ-carboxyl group to Glu residues, which are recycled back to the reduced active form by vitamin K epoxide reductase (VKOR; Figure 2). The vitamin K cycle is fundamentally a redox cycle that sustains reduced vitamin K.
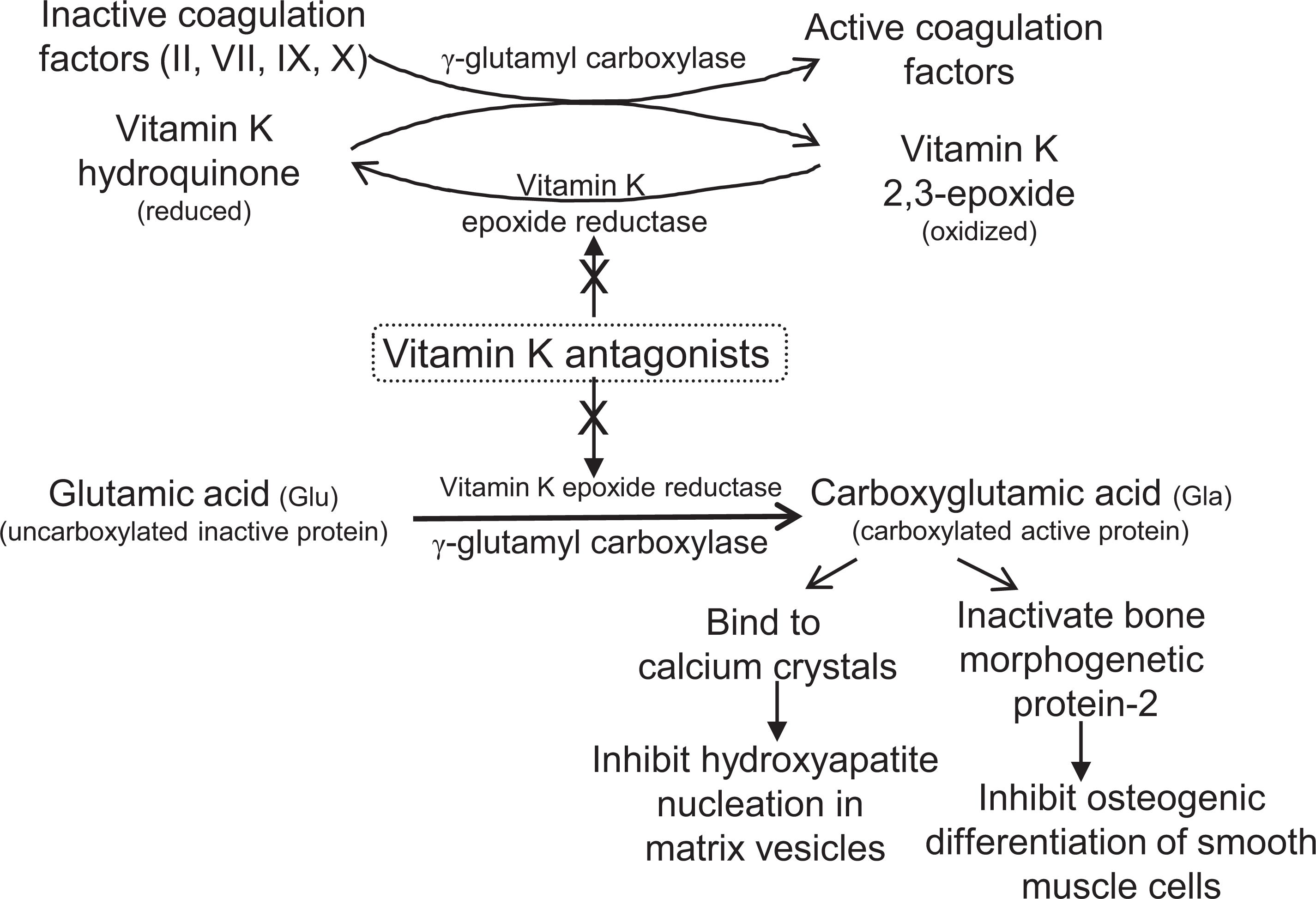
Pathways of vitamin K antagonist-induced calcification: Vitamin K hydroquinone is oxidized to vitamin K 2,3-epoxide form by vitamin K epoxide reductase. The oxidation of the vitamin K hydroquinone cycle is coupled to the carboxylation of vitamin K-dependent glutamic acid (Glu) residues. The γ-carboxylation reaction is catalyzed by γ-glutamyl carboxylase enzyme and requires the presence of a reduced form of vitamin K, resulting in the formation of glutamate (γ-carboxyglutamate acid [Gla]) residues and the epoxide form of vitamin K. Vitamin K antagonists, such as warfarin, inhibit coagulation factors by inhibiting the expression of vitamin K epoxide reductase. In addition, vitamin K antagonists inhibit formation of vitamin K-dependent γ-carboxylation of Glu residues, resulting in the accumulation of uncarboxylated proteins that lack calcification inhibitory capacity and promote vascular calcification. The γ-carboxylated Gla residues inhibit crystal growth, hydroxyapatite nucleation in vesicle membrane and smooth muscle cell transdifferentiation.
The γ-carboxylated vitamin K-dependent proteins bind to calcium ions and inhibit crystal growth in the extracellular matrix. The γ-carboxylated vitamin K-dependent proteins protect and maintain vascular calcification. Low levels of vitamin K lead to the production of undercarboxylated proteins and impair activation of Gla-containing calcification inhibitors. 33 A failure of γ-carboxylation leads to a cascade of events culminating in vascular calcification. A decrease in γ-carboxyglutamate protein or the ratio of active carboxylated to inactive uncarboxylated forms of γ-carboxyglutamate protein favor procalcific effects. In vitamin K deficiency or mutations in the γ-glutamyl carboxylase enzyme, undercarboxylated vitamin K-dependent proteins lead to the calcification process. 9,34 Thus, recycling the active form of vitamin K is necessary in activating vitamin K-dependent calcification modulator proteins.
Vitamin K-Dependent Modulators of Vascular Calcification
Vitamin K-dependent modulators of vascular calcification include inhibitory proteins, such as MGP, GRP, and growth arrest–specific 6 (Gas-6) protein, and stimulator proteins, such as osteocalcin.
Matrix Glu Protein
Matrix carboxyglutamate protein is a vitamin K-dependent extracellular matrix protein synthesized by vascular smooth muscle cells. 30 Matrix carboxyglutamate protein contains 5 Glu residues and 3 serine residues, requiring glutamate carboxylation and serine phosphorylation, respectively, to become fully functional to inhibit calcification. Matrix carboxyglutamate protein could be present as active isoform of dephosphorylated-carboxylated MGP and phosphorylated-carboxylated MGP, and inactive isoform of phosphorylated-uncarboxylated MGP (p-ucMGP) and dephosphorylated-uncarboxylated MGP (dp-ucMGP). The active form of γ-carboxyglutamate residue is formed by vitamin K-dependent carboxylation of Glu into matrix Gla protein. The γ-carboxylated active form of MGP binds to calcium ions with high affinity and inhibits calcium crystal deposition and growth of mineralized crystal. 31,33,34 It also inhibits matrix mineralization by regulating the apoptosis of smooth muscle cells. 4,26,35 In addition, γ-carboxylated MGP inhibits BMP-2-driven smooth muscle cell transdifferentiation to osteochondrogenic phenotypes. 19,25 In osteochondrogenic smooth muscle cells, MGP is downregulated and bone-associated proteins, such as ALP, are upregulated. Under such conditions, smooth muscle cells become susceptible to apoptosis, and secrete apoptotic bodies and extracellular vesicles, leading to calcification. 7
Active conformation of γ-carboxylated MGP inhibits the reprogramming of smooth muscle cells into osteoblast-like cells and prevents calcification. 35 The function of active MGP in suppressing calcification of arteries was demonstrated in inactivated MGP by treatment with warfarin, which showed progressive calcification. 9,36,37 In another study, MGP-deficient mice developed extensive calcification in the tunica media and died prematurely due to massive calcification, indicating the importance of activated MGP in preventing calcification. 38 Conversely, MGP overexpression reduces medial calcification and inhibits crystalline growth and stiffening of arteries due to calcium deposits. 26 In humans, mutations in the gene encoding the MGP also have been reported to induce concentric calcification. 39
Decreased activity of γ-glutamyl carboxylase leads to an undercarboxylated inactive form of MGP. When MGP is uncarboxylated, extracellular vesicles become nucleation sites, creating a procalcification milieu. 2 Downregulation of carboxylated MGP expression is associated with calcification of negatively charged cell membrane phospholipid remnants that have the capacity to nucleate calcium and phosphate. 9,33 A decrease in the ratio of carboxylated active MGP to uncarboxylated inactive conformation favors calcification. Non-fully γ-carboxylated MGP and reduced vitamin K-dependent carboxylation increase with vascular calcification. 40,41 Reduced vitamin K2 levels lead to increased plasma levels of uncarboxylated MGP and increase vulnerability to vascular calcification in dialysis patients. 31 Supplementation with vitamin K2 reduces circulating uncarboxylated MGP and inhibits warfarin-induced arterial calcification. 9,32 Thus, γ-carboxylated MGP decreases the number of calcification foci, acts as an inhibitor of deposition and crystallization of calcium, and inhibits calcification.
γ-Carboxyglutamate Acid–Rich Protein
Gla-rich protein is a vitamin K-dependent circulating protein, mainly expressed in cartilaginous tissues. 8 It has a high capacity to bind to calcium phosphate crystals and inhibits calcification. Like MGP, GRP is activated via posttranslational carboxylation of Glu residues in the presence of vitamin K. Gla-rich protein functions as a negative regulator of osteogenic differentiation by inhibiting expression of osteogenic genes and reducing differentiation of smooth muscle cells into osteoblast-like cells. 8 The γ-carboxylated GRP inhibits vascular mineralization by modulating crystal growth in the extracellular matrix and reducing hydroxyapatite nucleation in extracellular vesicle membrane. 8 In a procalcific milieu, upregulated GRP expression reduces calcification, but excess calcification stimuli may deplete vitamin K storage and hamper γ-carboxylation capacity, resulting in increased nonfunctional undercarboxylated protein forms. Gla-rich protein’s calcification inhibitory activity is dependent upon carboxylation, and undercarboxylated protein lacks calcification inhibitory capacity, promoting vascular mineralization.
Growth Arrest–Specific Gene 6 Protein
Growth arrest–specific-6 is a vitamin K-dependent protein expressed in a variety of cells, including smooth muscle cells, macrophages, and osteoblasts. 42 Like MGP, Gas-6 protein undergoes γ-carboxylation to convert into an active form that inhibit vascular calcification. Carboxylated Gas-6 stimulates the antiapoptotic protein Bcl-2, inhibits the pro-apoptotic proprotein caspase-3, and reduces apoptotic bodies. 2,43 It induces anticalcific effects by inhibiting smooth muscle cell apoptosis, facilitating phagocytosis of apoptotic cells and the resorption of hydroxyapatite. 43 The active form of carboxylated Gas-6 plays a role in regulating cell death, as well as in the migration of smooth muscle cells into areas of injury, and protects against calcium crystal formation.
Osteocalcin
Osteocalcin is a vitamin K-dependent matrix protein secreted by osteoblasts and smooth muscle cells that display an osteoblast-like phenotype. 6 Osteocalcin undergoes posttranslational γ-carboxylation to activate the protein’s calcium binding function, which has a high affinity for calcium and hydroxyapatite. Osteocalcin functions as a stimulator of differentiation and mineralization, upregulating Runx2, and ALP activity. 44 Under conditions of vitamin K deficiency or low activity of vitamin K-dependent carboxylase enzyme, osteocalcin can be undercarboxylated. In this form, it has less affinity to hydroxyapatite and loses its ability to bind to calcium deposits. Circulating γ-carboxylated osteocalcin promotes osteochondrogenic transdifferentiation of smooth muscle cells, leading to vascular calcification. 44 Osteocalcin promotes the incorporation of calcium into the extracellular matrix and enhances the mineralization process.
Vitamin K Inhibitors
Vitamin K antagonists inhibit enzymatic activation of γ-carboxylated Gla proteins and the recycling of vitamin K, thereby reducing the availability of the active conformation of the protein. 9,33 In the vitamin K cycle, hydroquinone of vitamin K is oxidized to vitamin K 2,3-epoxide, which is then converted to vitamin K hydroquinone by VKOR. Vitamin K epoxide reductase is needed for the synthesis of functional coagulation factors, such as factors II, VII, IX, and X and anticoagulant factors proteins C and S. Vitamin K antagonists inhibit the reduction of vitamin K to vitamin K hydroquinone by VKOR enzyme complex. This process leads to the accumulation of vitamin K 2,3-epoxide, thereby reducing the recycling of vitamin K epoxide. Vitamin K epoxide reductase inhibition leads to a reduced recycling of the active vitamin K form for carboxylation of Glu residues in blood-clotting proteins, resulting in bleeding.
Coumarin-derived drugs, such as warfarin, block the vitamin K cycle, leading to inhibition of vitamin K recycling into a functionally active form. Warfarin is a vitamin K antagonist that targets both vitamin K-dependent coagulation factors and vitamin K-dependent Gla-protein residues. 45 It inhibits γ-carboxylation of vitamin K-dependent proteins into an active conformation of Gla proteins, decreasing the ability of proteins to bind to calcium. 9,33
Inhibition of γ-carboxylation of Gla proteins by vitamin K antagonists promotes extracellular vesicles and apoptotic bodies. Reduced activity of vitamin K-dependent calcification inhibitor proteins during a critical period of embryonic developmental ossification causes calcification and skeletal abnormalities, leading to embryopathy, a complication of warfarin treatment in the first trimester of pregnancy. 46 Inhibition of the formation of an active carboxylated Gla protein by vitamin K antagonists leads to ectopic calcification of abnormal cartilage growth. Long-term exposure of smooth muscle cells to drugs that inhibit vitamin K leads to uncarboxylated inactive MGP conformation, which can induce osteo/chondrocytic differentiation of smooth muscle cells. Warfarin treatment of patients with coronary artery disease was reported to be associated with increased progression of coronary artery calcification. 36 Thus, vitamin K antagonists induce vascular calcification by inhibiting γ-carboxylation of vitamin K-dependent proteins, leading to reduced anticalcific capacity. 37,45,47
Vascular Calcification Markers
Several candidate surrogate markers of vitamin K-dependent and independent calcification factors have emerged, which need to be qualified as reliable predictors of vascular calcification. These include MGP, GRP, GAS-6, osteocalcin, fetuin-A, osteopontin, osteoprotegerin, pyrophosphates, and BMPs. This list is by no means exhaustive (Figure 3). The ratio of procalcific to anticalcific mediators determines whether calcification occurs independently of modification by vitamin K (Table 2). While a correlation between serum levels of markers and calcification has been proposed, whether the changes are attributed to causal association or to a compensatory process, the beneficial effects of modulating the proteins remain to be determined. 6,48–51

Vascular calcification inhibitors and activators: An imbalance in the expression of calcification inhibitors and promoters in the vascular wall could favor osteogenic differentiation of smooth muscle cells and activation of calcification. Calcification inducers activate differentiation of smooth muscle cells to chondrocyte/osteoblast-like cells by upregulation of transcription factors, such as Runx2, which incorporate calcium and phosphorus into extracellular vesicles to initiate mineralization and hydroxyapatite formation. Calcification inhibitory mediators inhibit hydroxyapatite deposition and smooth muscle cell differentiation into osteoblast. BMP-2 indicates bone morphogenetic protein; Gas-6, growth arrest–specific gene-6 protein; MGP, matrix γ-carboxyglutamate protein; MMPs, matrix metalloproteinases; Runx2, Runt-related transcription factor 2.
Circulating Biomarkers of Vascular Calcification.

Abbreviations: BMP-2, bone morphogenetic protein-2; Gal, γ-carboxyglutamate acid; GAS-6, growth arrest-specific 6 protein; MGP, matrix carboxyglutamate protein; RANKL, receptor activator of nuclear factor-κB ligand; TNF, tumor necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand; VSMC, vascular smooth muscle cell; OPG, osteoprotegerin.
Vitamin K-Dependent Biomarkers
Circulating levels of vitamin K-dependent γ-carboxylated MGP and GRP are inversely correlated with vascular calcification. 52,53 Plasma-undercarboxylated MGP and GRP could be potential markers for vascular calcification risk. 9 Low levels of carboxylated and phosphorylated MGP are associated with vascular calcification. 9,40 The nonfunctional forms of MGP, dp-ucMGP and p-ucMGP, have low affinity for calcium and high-circulating levels correlate with vascular calcification in patients treated with vitamin K antagonists. 40 Measurement of dp-ucMGP and p-ucMGP could be useful to monitor the effectiveness of vitamin K supplementation in hemodialyzed patients. 9,41 Serum levels of these markers could be useful in assessing the impact of long-term treatment with vitamin K antagonists on vascular calcification. Another vitamin K-dependent inhibitory protein, Gas-6, is inversely related to calcification, suggesting that circulating levels might be a biomarker for vascular calcification. 42 However, further studies are needed to support the role of Gas-6 in disease models of calcification. 43 A vitamin K-dependent procalcific protein, osteocalcin, positively correlates with differentiated smooth muscle cells into osteoblast-like cells. 6 Osteocalcin positively correlates with smooth muscle cells differentiated into osteoblast-like cells and with higher bone mineral density. 6 Whether circulating carboxylated osteocalcin concentration has utility as a marker of calcification processes needs further investigation.
Vitamin K-Independent Biomarkers
Fetuin-A (α2-Heremans Schmid glycoprotein) is a liver-derived circulating glycoprotein that inhibits hydroxyapatite formation and reduces crystal deposition of calcium and phosphate, independently of modification by vitamin K. 54,55 Fetuin-A inhibits vascular calcification by preventing extracellular vesicle calcium phosphate growth and reducing calcium-induced smooth muscle cell apoptosis. Low serum fetuin-A concentration is inversely associated with an increase in vascular calcification and reflects a procalcific milieu. 49,54,55 Serum levels of fetuin-A are inversely associated with vascular calcification; thus, circulating fetuin-A could indicate calcification. 51,56
Osteopontin is a phosphoprotein that inhibit calcification by blocking hydroxyapatite growth and crystal dissolution by inducing carbonic anhydrase expression, leading to acidification of the extracellular milieu. 57 Osteopontin deficiency results in vascular calcification in gene knock-out animal models indicating that it plays a role in inhibiting smooth muscle cell calcification by binding to mineralized crystal surfaces and blocking hydroxyapatite formation. 57 Reduced circulating levels of osteopontin are positively correlated with vascular calcification. 3,48
Osteoprotegerin is a secreted protein of the tumor necrosis factor (TNF) receptor superfamily and is an osteoclastogenesis inhibitory factor. 58 It acts as a decoy receptor for the receptor activator of nuclear factor-κB ligand (RANKL), a mediator of osteoblast maturation. Neutralization of RANKL by osteoprotegerin impairs osteoclastogenesis. 6,50,59 Osteoprotegerin prevents vascular calcification by blocking the matrix remodeling process, inhibiting ALP activity and neutralizing pro-apoptotic actions of TNF-related apoptosis-inducing ligand, which activate smooth muscle cell apoptosis. 59 Osteoprotegerin knock-out mice develop medial and subintimal calcification, indicating that osteoprotegerin plays a role in protecting against vascular calcification. 51,58 An imbalance in the osteoprotegerin/RANKL signaling pathway favors calcification, and thus, it could be used as a marker of vascular calcification. 48,50,53
Pyrophosphate acts by direct physicochemical inhibition of calcium phosphate precipitation and prevents hydroxyapatite formation. 60,61 Circulating inorganic pyrophosphate in blood plasma is negatively associated with calcification; thus, it serves as an endogenous inhibitor of hydroxyapatite formation. 33 Plasma concentration of pyrophosphate could be an indicator of the calcification process.
Bone morphogenetic protein-2 promotes the process of osteochondrogenic transdifferentiation of smooth muscle cells into osteoblast-like cells by upregulating the expression of Runx2. 19 It induces osteogenic phenotypic differentiation and enhances the release of apoptotic bodies from smooth muscle cells. 25 Inhibition of BMP-2 inhibits osteoblast differentiation and protects against vascular calcification. 25,26 Plasma BMP-2 levels are positively correlated with plaque progression and calcification. 62
Conclusions
Several studies have elucidated the complex mechanisms underlying vascular calcification, but many questions remain regarding the distinct pathways specific to the blood vessel wall. Unraveling these pathways will be helpful in developing new therapeutic strategies for preventing vascular calcification. Experimental evidence describes various players in the development of vascular calcification, including an imbalance of anticalcific and procalcific mediators, release of calcification-competent extracellular vesicles and apoptotic bodies, leading to increased osteogenic differentiation of smooth muscles cells and hydroxyapatite deposition. Inhibition of vitamin K-dependent proteins or conditions of vitamin K deficiency impair γ-carboxylation of proteins, resulting in the accumulation of undercarboxylated protein that lack calcification inhibitory capacity and, consequently, promote vascular mineralization.
The processes of vascular calcification are portrayed as resembling developmental bone formation, and their interrelatedness raises a challenge in designing selective therapeutic interventions that relieve one without impacting the other. An association of vascular calcification with reduced proteins, such as MGP, GRP, osteopontin, fetuin-A, among others, implicates these proteins as potential biomarkers. Whether these biomarkers can be employed to monitor calcification, and whether agents selectively targeting biomarkers of vascular calcification can reduce vascular calcification, deserves further investigation. Future efforts will likely focus on identifying key regulatory factors and interactions associated with smooth muscle cell differentiation and mineralization processes that can be specifically targeted to reducing calcification. Increased understanding of the medial smooth muscle cell switch into an osteogenic phenotype may uncover selective therapeutic targets that could limit the progression of vascular calcification.
Footnotes
Authors’ Note
This article reflects the views of the author and should not be construed to represent Food and Drug Administration’s views or policies.
Acknowledgments
The author would like to thank Joanne Berger for constructive criticism of the manuscript.
Author Contribution
Belay Tesfamariam contributed to conception, design, acquisition, analysis, and interpretation. He drafted the manuscript and critically revised the manuscript. He also gave final approval and agreed to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.