
Abstract
Microglia are a specialized type of neuroimmune cells that undergo morphological and molecular changes through multiple signaling pathways in response to pathological protein aggregates, neuronal death, tissue injury, or infections. Microglia express Trem2, which serves as a receptor for a multitude of ligands enhancing their phagocytic activity. Trem2 has emerged as a critical modulator of microglial activity, especially in many neurodegenerative disorders. Human TREM2 mutations are associated with an increased risk of developing Alzheimer disease (AD) and other neurodegenerative diseases. Trem2 plays dual roles in neuroinflammation and more specifically in disease-associated microglia. Most recent developments on the molecular mechanisms of Trem2, emphasizing its role in uptake and clearance of amyloid β (Aβ) aggregates and other tissue debris to help protect and preserve the brain, are encouraging. Although Trem2 normally stimulates defense mechanisms, its dysregulation can intensify inflammation, which poses major therapeutic challenges. Recent therapeutic approaches targeting Trem2 via agonistic antibodies and gene therapy methodologies present possible avenues for reducing the burden of neurodegenerative diseases. This review highlights the promise of Trem2 as a therapeutic target, especially for Aβ-associated AD, and calls for more mechanistic investigations to understand the context-specific role of microglial Trem2 in developing effective therapies against neurodegenerative diseases.
Microglia—Specialized Neuroimmune Cells
Microglia are the primary immune cells of the central nervous system (CNS) and play a pivotal role in maintaining neural health and homeostasis. Originating from the yolk-sac progenitors, these resident macrophages in the brain are critical for early brain development, synaptic pruning, and responding to injury or disease (Hickman and others 2018; Song and Colonna 2018). Microglia are highly dynamic, constantly surveying the neural environment for signs of injury and tissue damage or infection (Baik and others 2016). Upon encountering injury or pathogens, they quickly get activated and change their morphology and function in response to various chemical stimuli. This adaptability is critical for their roles in phagocytosis, secretion of cytokines, and modulation of inflammatory responses. In a healthy brain, microglia contribute to the regulation of neuronal activity, supporting synaptic plasticity and neurogenesis (Colonna and Butovsky 2017). However, in neurodegenerative diseases, such as Alzheimer disease (AD), Parkinson disease (PD), multiple sclerosis (MS), and amyotrophic lateral sclerosis (ALS), microglial activation may become dysregulated, leading to chronic inflammation and exacerbating neuronal damage (Hickman and others 2018; Song and Colonna 2018).
Microglia have been traditionally classified into activation states, categorized as proinflammatory (M1) and anti-inflammatory (M2) (Martinez and Gordon 2014; Michelucci and others 2009). However, recent advancements in the field have highlighted a more complex spectrum of microglial activation states, moving beyond the simplistic M1/M2 classification to include distinct microglial roles such as disease-associated microglia and specific responses to unique ligands like amyloid β (Aβ) and TDP-43, which demonstrate the dynamic and multifaceted nature of microglial functions in neurodegenerative processes (Paolicelli and others 2022). At the resting state, microglia have a ramified morphology with small nuclei. In neurodegenerative diseases, microglia can adopt an M1-like phenotype with chronic activation, leading to sustained release of proinflammatory cytokines and reactive oxygen species, thereby exacerbating neuronal damage. Microglia may also adopt an M2-like phenotype, aiding in tissue repair and debris clearance, thus exhibiting a neuronal protective role (Guo and others 2022) (Table 1). However, recent molecular analyses suggest that the M1 and M2 states are oversimplified and that microglial activation mechanisms may produce a variety of microglial subtypes with distinct features that are molecularly complex (Vidal-Itriago and others 2022). The recent identification of a unique microglial subtype in neurodegenerative diseases, termed disease-associated microglia (DAM) (Deczkowska and others 2018) or microglial neurodegenerative (MGnD) phenotype (Clayton and others 2021; Krasemann and others 2017), has been a significant development in the microglia field. The MGnD microglia not only are capable of phagocytosing dead or injured neurons or other cells, pathogens, and toxic substances but also play an important role in promoting inflammatory processes (Jiang and others 2014).
Microglia Subtypes in Healthy and Diseased Brain.
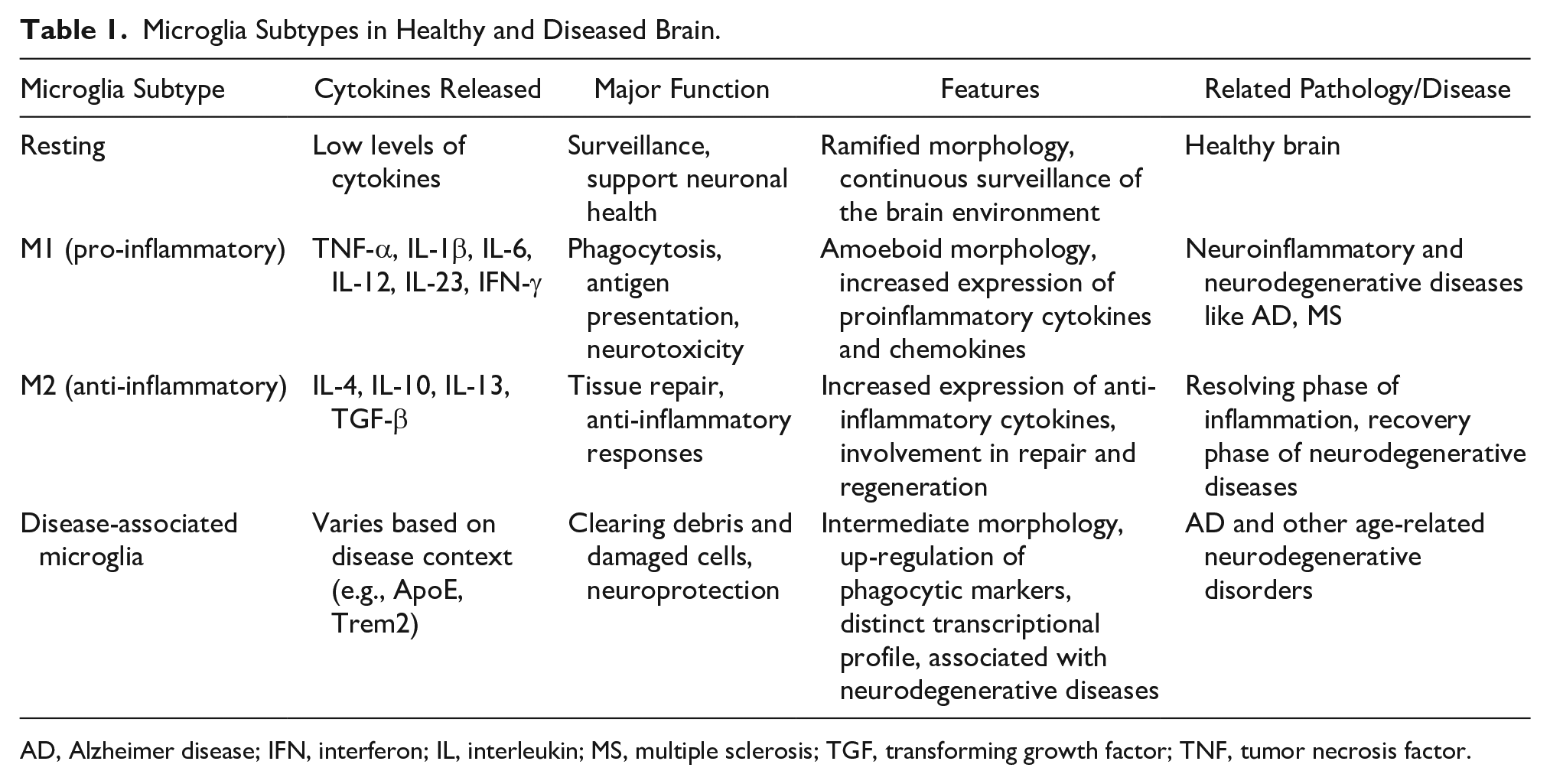
AD, Alzheimer disease; IFN, interferon; IL, interleukin; MS, multiple sclerosis; TGF, transforming growth factor; TNF, tumor necrosis factor.
Mechanistically, upon activation, microglia undergo metabolic changes to meet the biosynthetic and energetic demands of their altered functional states. This reprogramming includes shifts in glycolysis, oxidative phosphorylation, and lipid metabolism (Lauro and Limatola 2020). In the proinflammatory state, microglia rely more on glycolysis, as has been observed in peripheral macrophages (Lauro and Limatola 2020). In the context of neurodegenerative diseases, microglial activation is triggered by various signals, including damaged neurons, aggregated proteins (like Aβ or α-synuclein), and various pathogens. The activation of microglia occurs via several signaling pathways that include the nuclear factor (NF)–κB pathway, inflammasomes, and various receptor-mediated pathways (Wangler and Godbout 2023), which help regulate the production of cytokines, chemokines, and other cell signaling mediators.
Given the pivotal role of microglia in neuronal homeostasis and neurodegenerative diseases, these cells have become a focal point in understanding the molecular mechanism where they interface with neurodegenerative disorders like AD, PD, MS, and ALS. Their dual functions as both the protectors and potential contributors to neuronal pathology underscore the complexity and the critical need for in-depth investigations into their multifaceted roles in CNS homeostasis.
Microglia Express a Major Regulator for Their Activation
Microglia express the triggering receptor expressed on myeloid cells 2 (Trem2) protein, which has emerged as a critical immunological signaling receptor in response to various pathologies. Trem2 interacts with a multitude of ligands that are frequently linked to tissue injury (Deczkowska and others 2020). While the activity of Trem2 is constrained under normal cellular circumstances, it assumes a crucial role for identifying and minimizing tissue damage during pathogenesis. Currently, enormous efforts are under way to exploit Trem2 as a therapeutic option given its pivotal role in microglia and their activation. This review attempts to provide an overview of the current state of Trem2 signaling and how it interfaces with many diseases, specifically neurological disorders. It delves into the molecular aspects of Trem2 and how it affects microglial cells, with particular attention on how it affects metabolism and phagocytosis in the brain, improves cell survival, and reduces neuroinflammation.
Discovery of the Trem2 Gene
The human TREM2 gene is located on chromosome 6p21.1, and this chromosomal region is known to carry several genes that are important for immune system function. In mice, the Trem2 gene is located on chromosome 17, which corresponds to a part of the human chromosome 6, reflecting a degree of genetic conservation and chromosomal synteny between the species. Trem2 was first identified in the early 2000s as part of the TREM family of receptors, which are expressed on the surface of immune cells and involved in modulating macrophage immune responses. Bouchon et al. identified Trem2 as a receptor expressed on monocytes and macrophages (Bouchon and others 2000; Bouchon and others 2001), suggesting its involvement in innate immunity. These seminal studies laid the foundation for understanding Trem2’s role in immune modulation and regulation. The role of Trem2 took a significant turn when it was discovered that Trem2 is expressed in the brain, particularly in the microglia, the CNS resident immune cells. In 2002, Schmid et al. (Schmid and others 2002) demonstrated the expression of Trem2 in microglia, suggesting a potential role in neuroinflammatory processes. This was a pivotal moment, linking Trem2 to neuronal function and neurodegenerative diseases. The functional association between human TREM2 and neurodegenerative diseases became clearer with subsequent studies. In 2013, landmark studies by both Guerreiro et al. (Guerreiro and Hardy 2013) and Jonsson et al. (Jonsson and others 2013) found a rare mutation that changed arginine at position 47 to histidine (R47H) in the human TREM2 gene, which significantly increased the risk of AD. Both discoveries, reported in the New England Journal of Medicine, were crucial in establishing Trem2 as a key player in AD pathology. Subsequent studies confirmed an additional mutation R62H along with R47H as the highest frequency changes in familiar AD cases (Jin and others 2014).
Further studies addressed the mechanisms by which Trem2 influences and impacts neurodegenerative processes. In 2017, Ulland et al. (Ulland and others 2017) reported that TREM2 mutations impair the functions of microglia, contributing to AD pathology providing details in understanding the functional aspects of Trem2 in disease contexts. The most recent studies continue to unravel the complexities of Trem2 in neurodegeneration. For instance, in 2017, a study by Krasemann et al. (Krasemann and others 2017) provided insights into how Trem2 signaling in microglia influences the immune response in the brain, further elucidating its role in neuroinflammatory conditions associated with the AD. The direct involvement of Trem2 in AD pathogenesis was notably demonstrated by findings that Trem2 directly interacts with Aβ (Lessard and others 2018; Zhao and others 2018), influencing the trafficking and internalization of Aβ aggregates into microglia. This discovery highlights Trem2’s critical role in AD’s progression by modulating key pathological molecules. Thus, the initial discovery and subsequent studies on Trem2 (summarized in Fig. 1) have been instrumental in understanding immune regulation and neurodegenerative diseases. From its initial identification in immune cells to its crucial role in microglia and AD, the Trem2 receptor has emerged as a key protein of interest in both immunology and neurobiology. The understanding of Trem2 as a key regulator in immune cell metabolism has become a driving force in most recent investigations for neurological disorders, particularly in AD, providing avenues to target Trem2 for therapeutic benefits.
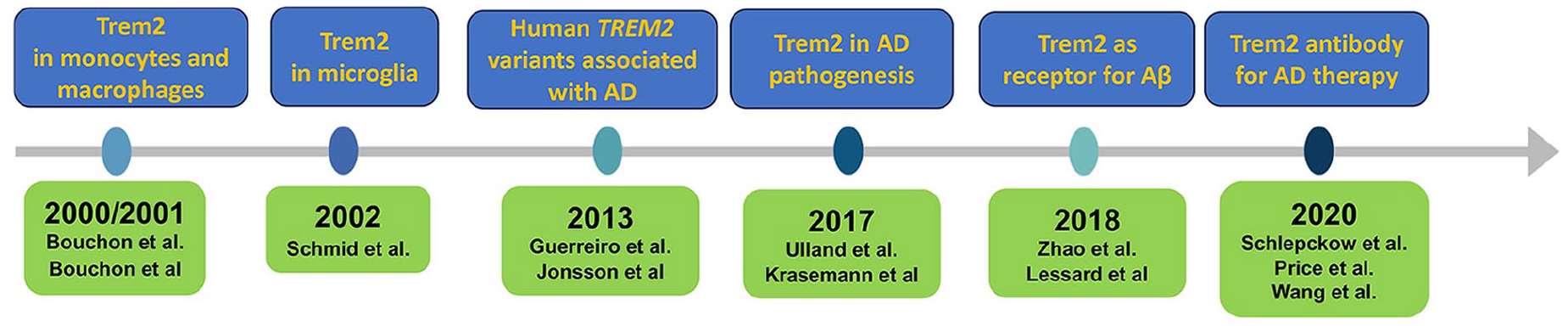
Historical milestones in microglia-Trem2 research. Schematic illustration of the key milestone events from various research groups that contributed to the discovery of Trem2 in microglia and its importance in Alzheimer disease research.
Basic Molecular Structure and Functions of Trem2
The Trem2 protein is a single-pass transmembrane receptor belonging to the immunoglobulin superfamily (Ig-SF) of proteins. It consists of an extracellular V-type immunoglobulin (IgV) domain, a long stalk, a single transmembrane helix that interacts with the adaptor protein DAP12 via lysine–aspartic acid interaction, and a short cytosolic tail containing an immunoreceptor tyrosine-based activation motif (ITAM) (Lanier and others 1998) (Fig. 2). The binding of DAP12 is crucial for both Trem2 membrane stabilization and downstream signaling (Peng and others 2010). The identified natural ligands of Trem2 include various phospholipids (Wang and others 2015), glycolipids, lipidated particles (e.g., high-density lipoprotein and low-density lipoprotein) (Shirotani and others 2019), and lipoproteins like APOE and CLU/APOJ (Yeh and others 2016). It can also bind to Aβ oligomers (Zhao and others 2018).
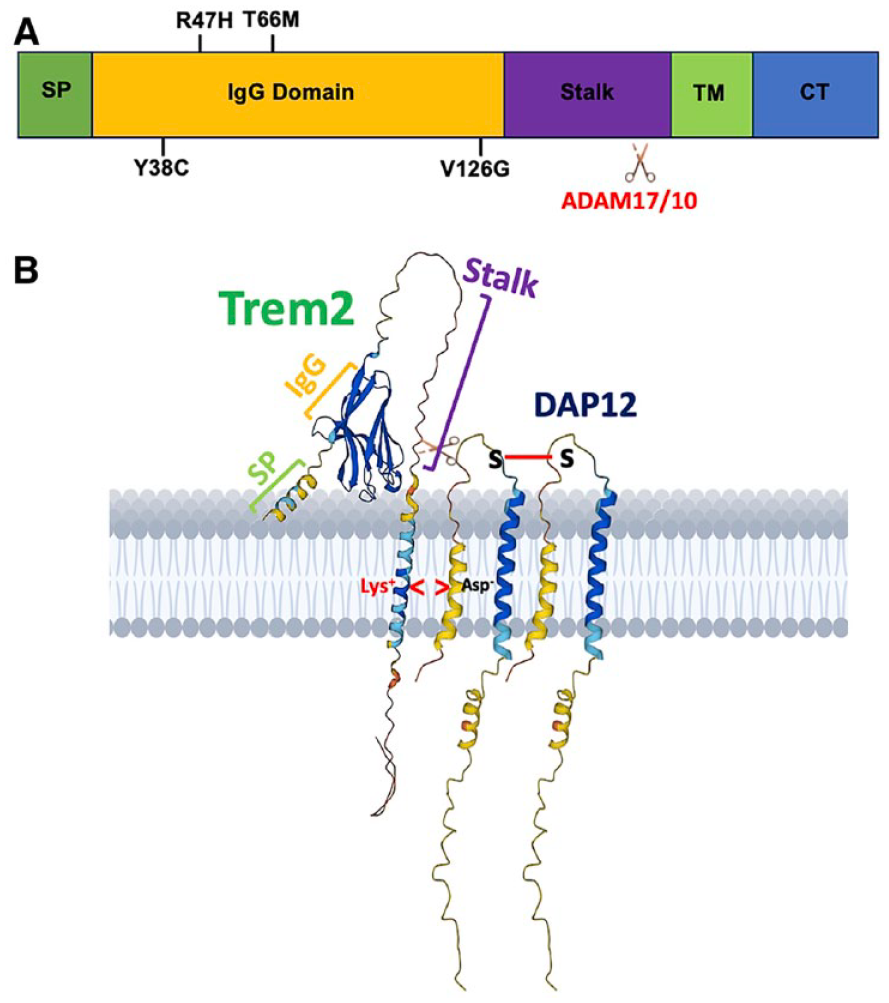
Domain structure of the Trem2 protein and its association with Alzheimer disease (AD) risk. (A) Human Trem2 protein domain structure: this schematic illustrates the structural domains of the human Trem2 protein, including the signal peptide (SP), transmembrane domain (TM), and C-terminal tail (CT). Notable are the common variants associated with AD risk and the cleavage sites for ADAM17/10, which are crucial for Trem2’s functional modulation in the disease context. (B) Membrane-bound three-dimensional (3D) structure of Trem2-DAP12 complex: derived from AlphaFold (ID: Q9NZC2), this representation showcases the 3D conformation of Trem2 as it anchors to the cell membrane through a transmembrane helix. TREM2’s interaction with DAP12 (AlphaFold ID: O43914) is highlighted, showing electrostatic interactions (involving lysine and arginine residues) within the transmembrane domains. Additionally, the dimerization of two DAP12 proteins through a disulfide bond is depicted, emphasizing the complex’s structural integrity and signaling capability.
Another critical aspect of Trem2 is the shedding of its ectodomain, facilitated by ADAM10/17 at histidine 157 (H157) in the stalk region, which leads to the formation of soluble Trem2 (sTrem2) (Jay and others 2017b). sTrem2 is found in the cerebrospinal fluid (CSF), plasma, and brain tissues (Morenas-Rodriguez and others 2022). It is thought to have a variety of functions, including the regulation of inflammation, phagocytosis, and synaptic plasticity (Biel and others 2023; Weber and others 2022). In general, sTREM2 can act in a neuroprotective manner. It has been shown to promote the survival and proliferation of microglia, enhancing their phagocytic capabilities to clear Aβ plaques, a hallmark of AD pathology (Ewers and others 2019; Suarez-Calvet and others 2016). Mechanistically, sTrem2 has been shown to interact with APOE (Yeh and others 2016), an important membrane-bound lipid regulator and a major genetic risk factor for AD. These interactions modulate microglial functions, influencing the inflammatory response and potentially affecting the progression of neurodegeneration (Shi and Holtzman 2018). sTREM2 may also influence the complement system, which plays a role in marking synapses for clearance by microglia during inflammatory responses (Fraser and others 2010; Scott-Hewitt and others 2020). This interaction is essential for the regulation of synaptic pruning, which can be aberrant in AD, leading to synaptic loss and associated cognitive decline (Hammond and others 2018). The combination of specific molecular domains allows Trem2 to function as a receptor on the surface of immune cells like microglia, playing a vital role in immune surveillance, phagocytosis of cellular debris, and regulation of inflammatory responses. The involvement of Trem2 in neurodegenerative diseases such as AD is collectively linked to these functions, particularly its role in the clearance of Aβ plaques and the regulation of neuroinflammation.
Diverse Ligand Interactions of Trem2
As presented above, Trem2 has been reported to bind to a variety of ligands, reflecting its role in immune responses and neurodegeneration. Specifically, it binds to ApoE, a key player in AD, and this interaction is crucial for modulating microglial functions, such as phagocytosis of apoptotic neurons (Atagi and others 2015; Bailey and others 2015; Yeh and others 2016). Furthermore, Trem2 directly engages with Aβ oligomers with nanomolar affinity, which bears significant implications for AD pathology (Lessard and others 2018; Zhao and others 2018). Functionally, the ligand-binding capacity of Trem2 extends beyond these proteins. Notably, clusterin (CLU), also known as apolipoprotein J (ApoJ), has been identified as another vital ligand for Trem2 (Yeh and others 2016). This interaction is especially relevant given CLU’s role in modulating inflammation and its overexpression in various neurodegenerative conditions. Trem2 also associates with anionic lipids, lipoproteins, and other polyvalent ligands (Belsare and others 2022; Ibach and others 2021; Kober and others 2016; Wang and others 2015). Specific phospholipids like phosphatidylserine and phosphatidylethanolamine are recognized by Trem2, particularly during the clearance of apoptotic cells (Shirotani and others 2019) . Additionally, Trem2 interacts with TDP-43, a protein associated with neurodegeneration, and mediates neuroprotection in TDP-43–related neurodegenerative conditions (Zheng and others 2017). Thus, a multitude of ligands play a significant role in Trem2’s function in immune regulation, neuroprotection, and the pathogenesis of neurodegenerative diseases. The diversity of these ligands underscores the multifaceted role of Trem2 in the neuroimmune modulation in the CNS and may involve ligand-specific Trem2-binding proteins that remain to be identified.
General Signaling Pathways Downstream of Trem2
The internal cellular signaling pathways influenced by Trem2 are integral to understanding its role in immune regulation and neurodegenerative diseases. Trem2 engages in signaling pathways that are crucial for microglial activation, survival, and phagocytic functions (Olufunmilayo and Holsinger 2022). Upon binding to its ligands, Trem2 activates downstream signaling cascades involving the adapter protein DAP12. This activation leads to the phosphorylation of ITAM motifs on DAP12, subsequently triggering a cascade of intracellular events (Fig. 2). These events include the activation of spleen tyrosine kinase (Syk) and phospholipase C (PLCγ2), which are pivotal in calcium signaling and cytoskeletal rearrangement (Deczkowska and others 2020). This signaling cascade enhances microglial survival and promotes phagocytic activity, crucial for clearing cellular debris and any misfolded proteins in the brain (Yao and others 2019). Moreover, the internal cellular signaling pathways affected by Trem2 play a crucial role in balancing proinflammatory and anti-inflammatory responses in microglia, processes that are vital for normal brain health and function (Liu and others 2020). When Trem2 is activated, it influences key signaling pathways such as NF-κB and Mitogen-activated protein kinases (MAPK), which are central to the regulation of inflammatory responses. Activation of NF-κB leads to the production of proinflammatory cytokines, while its regulation or inhibition can help mitigate excessive inflammation (Yao and others 2019). Concurrently, Trem2 signaling can activate anti-inflammatory pathways, likely through the induction of molecules like interleukin 10 and transforming growth factor β, which are known to suppress inflammatory responses and promote tissue repair (Cai and others 2022). This dual action of Trem2, in both promoting and restraining inflammation, exemplifies its role in maintaining a delicate balance between pro- and anti-inflammatory states. By modulating these cellular signaling pathways, Trem2 helps ensure that microglial activation is appropriate to the context, preventing chronic inflammation that can lead to neuronal damage, while still allowing for effective immune responses and debris clearance in the brain. Understanding and harnessing this balancing act of Trem2 is key to developing targeted therapies for neurodegenerative diseases where dysregulation of neuroinflammation is a key feature.
Trem2 and Microglial Functional States
Trem2 is predominantly expressed in the immune cells of myeloid lineage, including monocytes, macrophages, dendritic cells, and microglia, with its distribution and expression varying across different tissues and cell types (Rodriguez-Gomez and others 2020). In the CNS, Trem2 expression is most notably observed in microglia. Unlike peripheral macrophages, microglia are unique to the brain and spinal cord and are critical for maintaining CNS homeostasis.
Influence of Trem2 on Microglial Activation States
Microglia, the primary immune cells in the brain, play a crucial role in maintaining neural homeostasis, immune surveillance, and responding to injury or disease. The classical M1/M2 classification system for microglia, which categorizes these cells into either proinflammatory (M1) or anti-inflammatory (M2) states, has long served as a fundamental framework in neuroimmunology (Martinez and Gordon 2014; Michelucci and others 2009). However, recent advances in molecular biology and single-cell technologies have revealed that this binary classification is overly simplistic and inadequate for capturing the diverse and dynamic roles that microglia play in the CNS. Studies now show that microglia exhibit a spectrum of activation states that are influenced by a complex interplay of environmental signals, disease contexts, and temporal changes. In 2022, Paolicelli et al. published a landmark review article discussing the latest research advances about microglia, particularly their role in neurodegenerative diseases, and provided a refined classification of microglial states that better captures the complex and dynamic nature of these cells in various CNS disease contexts (Paolicelli and others 2022). These insights have necessitated a reevaluation of the traditional model, leading to the proposal of new classification system that reflects the multifaceted nature of microglial functions. Here, we adopted the discussion in the review article and summarized the role of Trem2 in this newly defined microglia-homeostatic status in neurodegenerative diseases.
Homeostatic microglia
In the homeostatic state, microglia perform surveillance and maintenance duties within the CNS, such as clearing metabolic waste and synaptic pruning. While TREM2 expression in homeostatic microglia is relatively low compared to activated states, it still plays a subtle role in maintaining the microglial sensitivity to changes in their environment (Hou and others 2022). Low-level TREM2 signaling in these cells may aid in the basal phagocytic activity and the processing of apoptotic cells, thereby preventing unnecessary immune activation and maintaining CNS homeostasis (Mazaheri and others 2017; McQuade and others 2020).
Disease-associated activation state microglia (DAM)
This group of microglia is characterized by a distinct gene expression profile that includes up-regulation of neurodegenerative disease–related genes. These microglia are thought to be initially protective, responding to neuronal damage by clearing debris and limiting inflammation (Keren-Shaul and others 2017). Trem2 is significantly implicated in the regulation of the DAM state, which will be discussed in detail in the following sections.
Interferon-responsive/major histocompatibility complex II high microglia
In this activation state, microglia respond to viral infections or other strong immune stimuli that activate pathways related to antigen presentation and more robust inflammatory responses. TREM2’s role in this state is complex; while not directly involved in the interferon response itself, TREM2 signaling can modulate the overall reactivity of these cells. For example, TREM2 can influence the lipid metabolism within microglia (Hou and others 2022), which is crucial for the synthesis of membranes needed for the formation of major histocompatibility complex class II complexes and the presentation of antigens. Moreover, TREM2 may help to balance the proinflammatory signals induced by interferon signaling, thereby preventing excessive microglial activation that could lead to bystander neuronal damage.
Trem2 in Microglial Phagocytic Activity
One of the key functions of microglia is the phagocytosis of cellular debris, pathogens, and apoptotic cells. Trem2 significantly influences this aspect of microglial function. Upon ligand binding, DAP12 phosphorylation leads to a downstream signaling cascade through Syk, PLCγ2, diacylglycerol (DAG), IP3/Akt, and mammalian target of rapamycin (mTOR) pathways, which in turn induces calcium release from intracellular stores (see review by Deczkowska and others 2020). Elevated calcium levels, along with DAG, activate protein kinase C (PKC) and other kinases, facilitating actin cytoskeletal rearrangements essential for phagocytic cup formation and engulfment of targets (Jairaman and others 2022). This signaling cascade also influences gene expression related to phagocytosis and inflammation, potentially involving transcription factors like NF-κB and MAPK pathways, thus regulating microglial responses crucial for debris clearance in the brain (Liu and others 2022). Understanding this intricate signaling pathway is vital for developing therapeutic strategies targeting microglial functions in neurodegenerative diseases.
Trem2 plays a critical role in microglial function beyond DAP12 signaling. It facilitates the recognition and attachment to a spectrum of ligands, including various lipids, lipoproteins, and other polyvalent ligands, ultimately promoting their clearance from the pathological brain (Belsare and others 2022; Ibach and others 2021; Kober and others 2016; Wang and others 2015). Recent studies have revealed that Trem2 interacts directly with Aβ, facilitating its clearance (Zhao and others 2018). Microglia that express TREM2 have an enhanced ability to bind to and phagocytose Aβ aggregate; however, loss of Trem2 function results in reduced phagocytosis of Aβ, exacerbating plaque accumulation and contributing to increased neuroinflammation and neurodegeneration. Trem2 also interacts with TDP-43 and facilitates the clearances of TDP-43 aggregates, which is implicated in ALS and frontotemporal dementia (Xie and others 2022a; Xie and others 2022b). By promoting the phagocytosis of TDP-43, TREM2 may also help reduce the proinflammatory stimuli associated with aggregated TDP-43, potentially decelerating the progression of disease.
In MS, a CNS demyelinating disease, Trem2 was highly expressed on myelin-laden phagocytes in active lesions. Trem2 promotes microglial survival, proliferation, and phagocytic activity, enhancing the clearance of myelin debris. This is crucial for remyelination and recovery in MS (Cignarella and others 2020). Similarly, in a leukoencephalopathy mouse model, Trem2 elevation was associated with demyelination in corpus callosum. The absence of Trem2 attenuated myelin pathology, suggesting that Trem2 contributes to microglial dys-homeostasis and potentially plays a role in demyelinating pathologies (Biundo and others 2023). Trem2 activation has also been linked to promoting remyelination, further underscoring its role in debris clearance and recovery processes in CNS myelination (Wood 2020). These studies collectively demonstrate the multifaceted role of Trem2 in microglial debris clearance, highlighting its significance in neurological diseases such as MS and leukoencephalopathy and in the context of neuronal death and remyelination.
TREM2 in Microglia Proliferation
Trem2 is pivotal not only for the phagocytic activity of microglia but also for their proliferation and survival, particularly in the context of neurodegenerative diseases. Trem2 is expressed on the surface of microglia, where it plays a crucial role in mediating the proliferation response of these cells under stress conditions or during disease progression (Painter and others 2015). As we discussed above, Trem2 functions by sensing and binding to various ligands, including damaged neurons and apoptotic cells, which not only promote phagocytic activity but also stimulate microglial proliferation. Upon ligand binding, Trem2 activates the ITAM-Syk signaling pathway. This pathway is critical for the activation of downstream signaling molecules such as PI3K/Akt, which enhances cell survival and promotes proliferation through the inhibition of apoptotic pathways and stimulation of cell growth signals. Akt stimulates cell growth and survival by activating mTOR (Saxton and Sabatini 2017). mTOR is a critical regulator of protein synthesis and cell growth and might also promote the expression of Trem2 on a feedback loop under the AD pathogenesis condition (Shi and others 2022). Additionally, TREM2 supports microglial survival by engaging the Wnt/β-catenin signaling pathway, particularly through posttranslational modifications that stabilize β-catenin (Zheng and others 2017). This mechanism offers insights into how diminished TREM2 function could compromise microglial viability, potentially exacerbating AD pathology. Understanding these pathways provides insights into how microglia respond to neurodegenerative challenges and opens avenues for therapeutic interventions that can finely tune microglial activities to harness their protective functions while mitigating harmful overactivation.
TREM2 in AD and Other Neurodegenerative Diseases
Trem2 has emerged as a key regulator of microglial functions, playing significant roles in neuroinflammation, phagocytic clearance of debris, and neuroprotective responses. Consequently, mutations in the TREM2 gene have been linked to increased susceptibility to AD. This connection highlights its critical role in the long-term maintenance of neural health and homeostasis.
TREM2 Variants Increase AD Risk
AD is a complex neurodegenerative disorder characterized by cognitive decline and memory loss. Recent research has increasingly focused on the genetic factors contributing to AD risk, with particular attention on variants of the TREM2 gene that have been linked to altered microglial function and increased susceptibility to AD (Zhou and others 2019) (summarized in Table 2). In 2013, two independent studies by Jonsson et al. (Jonsson and others 2013) and Guerreiro et al. (Guerreiro and Hardy 2013) identified a rare variant, R47H in human TREM2 gene, which significantly increased the risk of developing AD. This discovery marked a significant shift in understanding the genetic landscape of AD, beyond the well-known APOE ε4 allele (Strittmatter and others 1993). Additional studies have also shown its clinical relevance in other populations (Rikos and others 2022). The connection between TREM2 variants and AD risk was first highlighted in genome-wide association studies. A series of follow-up studies using transgenic mouse models showed the clinical relevance of Trem2 mutations (Sayed and others 2021; Tran and others 2023). A recent study discusses the development of a new mouse model, named LOAD, which expresses humanized risk factors for late-onset AD, including APOE ε4, Trem2 R47H, and Aβ (Kotredes and others 2021). This model aims to better mimic the clinical heterogeneity observed in LOAD patients and could be instrumental in understanding the disease and developing therapies. Penney et al. (Penney and others 2024) used gene editing and stem cell models to investigate the effects of the TREM2 R47H/+ mutation on human-induced pluripotent stem cell–derived microglia. It was observed that the mutation leads to a proinflammatory gene expression signature and impairs microglial movement and uptake of substrates. This study also revealed that mouse brains transplanted with Trem2 R47H/+ microglia exhibited reduced synaptic density, suggesting a detrimental effect on microglia linked to AD. Another mutation in Trem2, T66M variant was first described in a Turkish patient with frontotemporal dementia (FTD) (Guerreiro and others 2013b). While the R47H mutation increases AD risk, loss-of-function mutations like TREM2 T66M result in more severe forms of neurodegeneration. Current evidence indicates that the presence of the T66M variant in individuals is associated with significantly reduced concentrations of soluble Trem2 (sTrem2) in the cerebrospinal fluid (Henjum and others 2016; Heslegrave and others 2016; Piccio and others 2016). Recent studies have used gene editing and induced Pluripotent Stem Cells (iPSC) stem cell models to further investigate the effects of the Trem2 variants on microglial functions (Deming and others 2019).
TREM2 Variants Associated with Increased AD Risk and Other Neurodegenerative Diseases.
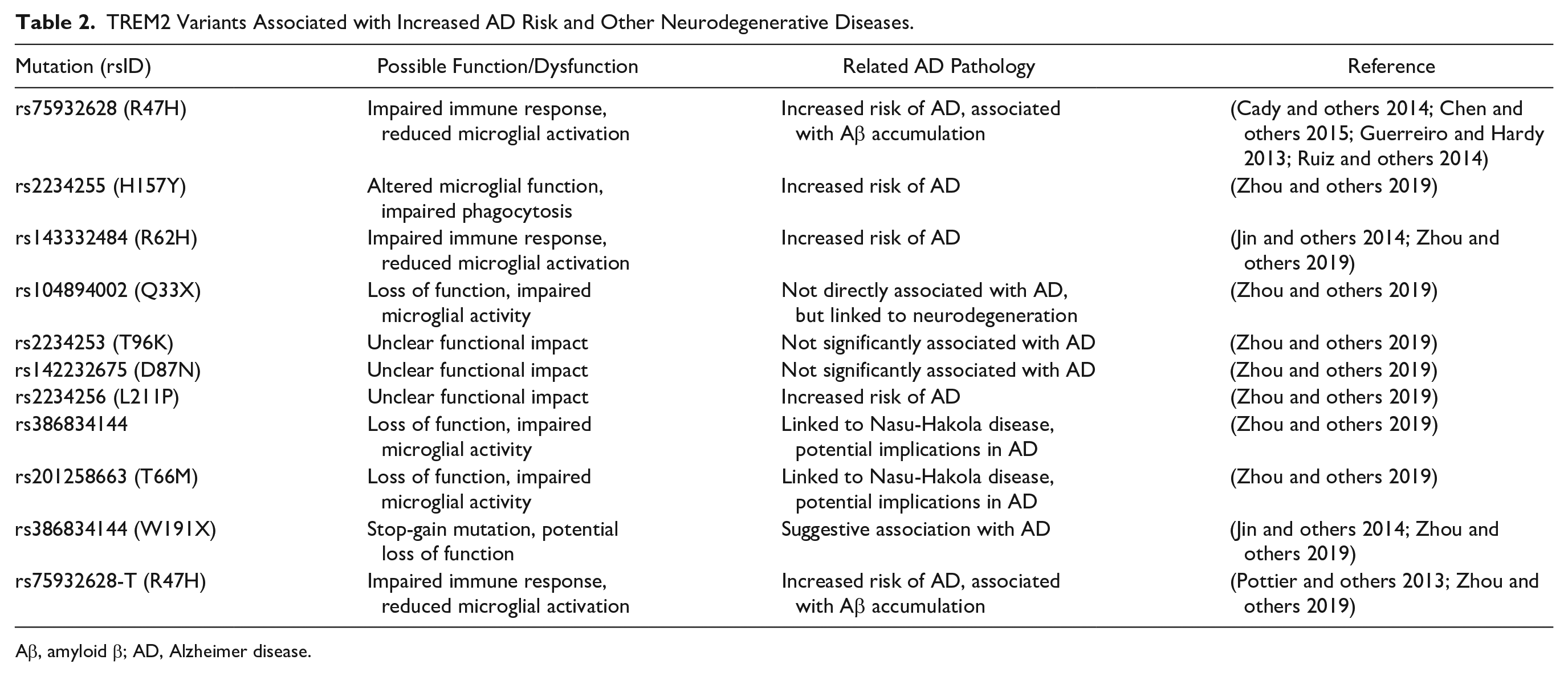
Aβ, amyloid β; AD, Alzheimer disease.
Furthermore, TREM2 variants have been studied in relation to PD and ALS, although the associations are less clear compared to AD and FTD. In PD, Trem2 expression levels have been observed to change, potentially influencing the microglial response to neuronal damage (Dardiotis and others 2021; Huang and others 2024). In ALS, while direct links to TREM2 mutations are not well established, alterations in Trem2 expression and function might contribute to the observed neuroinflammation and microglial activation in the disease (Jerico and others 2023; Siokas and others 2021).
Mechanisms Linking TREM2 Variants to AD
Impaired microglial function
TREM2 variants, notably R47H, result in dysfunctional microglia that are less efficient in clearing Aβ plaques, a hallmark of AD pathology. The impaired phagocytic activity leads to plaque accumulation, triggering neuroinflammation and neuronal damage (Meilandt and others 2020). Interestingly, the effects of Trem2-mediated microglial activation on Aβ pathology appears to be stage dependent. In early-stage AD, Trem2 deficiency is associated with a reduced Aβ load (Jay and others 2015), whereas in the late stage, an increase in Aβ load has been observed (Jay and others 2017a). Importantly, overexpression of Trem2 promotes secretion of inflammatory factors that potentially facilitate plaque seeding in the initial stages of Aβ accumulation (Ulland and others 2017). This is observed in mouse models with increased expression of cytokines and chemokines in the forebrain (Hou and others 2022). However, the roles for Trem2 in pure tauopathy mouse models appear to be more complex and somewhat contradictory (Bemiller and others 2017; Sayed and others 2018). Some studies have shown that Trem2 deficiency led to less microgliosis, astrogliosis, and expression of markers of DAMs and inflammation, leading to reduced hippocampal atrophy (Gratuze and others 2020). In summary, Trem2 plays a multifaceted role in AD pathology, with its impact varying across different stages of the disease and in different pathological contexts (summarized in Fig. 3), highlighting the complex interplay between microglial function and neurodegeneration in AD.
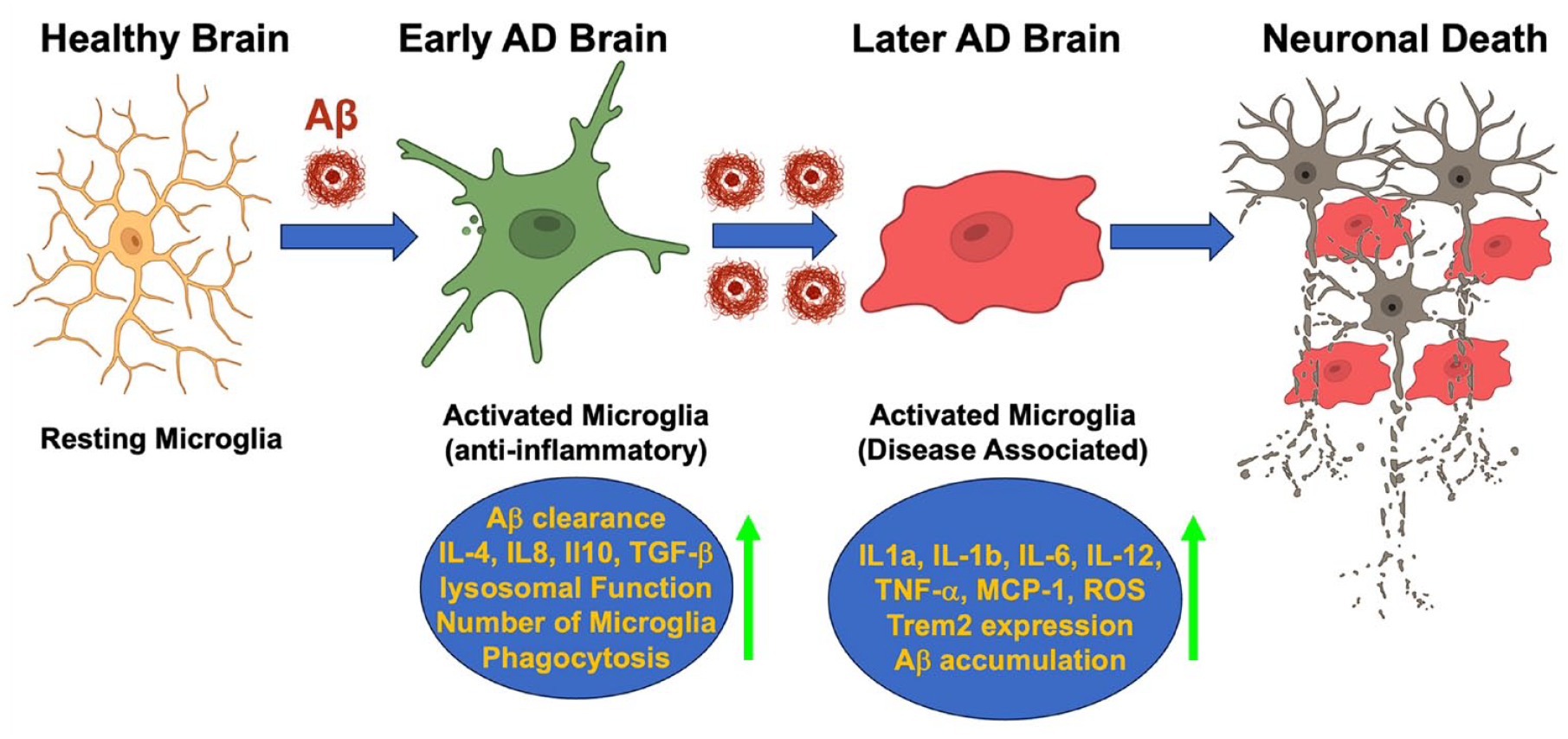
Homeostasis of microglia during Alzheimer disease (AD) pathogenesis. At the earlier stages of AD, microglia are activated in response to the expression of Aβ and reprogramed to increase amyloid β (Aβ) uptake and subsequently clearance, with the release of multiple cytokines contributing to the homeostatic maintenance of CNS. However, with the AD progression and accumulation of Aβ plaques, excessive activation of microglia would release excessive proinflammatory factors and increased reactive oxygen species molecules, to compromise neurons and their synapses, leading to neuronal death.
Interactions between Trem2 and Aβ
Trem2 deficiency led to impairment in Aβ degradation, and microglial signaling was altered in response to Aβ stimulation (Ulland and others 2017). Mechanistically, Trem2 has been shown to directly bind to Aβ oligomers, mediating various microglial functions critical in AD pathogenesis (Zhao and others 2018). These studies were the first to demonstrate that Trem2 bound to Aβ with nanomolar affinity. Additional studies provided evidence of high-affinity interactions between Aβ oligomers and Trem2 and changes in microglial signal transduction, underscoring the significance of Trem2 in AD pathology (Lessard and others 2018). These interactions are characterized by very slow dissociation rates between Trem2 and Aβ. Interestingly, TREM2 variants, specifically the R47H and R62H, which are contributing to AD pathology, while binding Aβ with equivalent affinity, showed a loss of function in NFAT signaling and reduced Aβ internalization (Lessard and others 2018). In addition, these studies suggest that high-affinity binding of Trem2 to Aβ oligomers can block interactions with other Trem2 ligands, such as ApoE. These findings highlight the complex role of Trem2 in neuroinflammatory processes in AD and potentially other neurodegenerative disorders.
Effect on synaptic and neuronal loss
Latest studies suggest that Trem2 variants have an impact on microglia-neuron communication (Tagliatti and others 2024). Trem2 is well documented to be involved in microglia-mediated synaptic pruning (Filipello and others 2018; Zhong and others 2023). While synaptic pruning is considered a normal process in a healthy brain, in AD, this process becomes dysregulated. Abnormal Trem2 function could lead to excessive or insufficient pruning, disrupting neural networks, which is thought to contribute to cognitive decline in AD (Filipello and others 2018). In addition, Trem2 influences the bioenergetics of neurons (Tagliatti and others 2024), and in the context of AD, impaired Trem2 function could affect neuronal energy metabolism, exacerbating the energy deficits commonly observed in AD brains (Qu and Li 2020). These factors collectively contribute to the neural network disruptions and energy deficits that underlie the cognitive decline in AD, underscoring the importance of Trem2 in maintaining neural health and function.
Influence on tau hyperphosphorylation and aggregation
Trem2 variants have also been associated with the modulation of tau protein hyperphosphorylation and aggregation, processes central to AD development (Bemiller and others 2017). Abnormal tau aggregation leads to the formation of neurofibrillary tangles, another key pathological feature of AD. Studies have shown that in the CSF from patients with AD, the soluble form of Trem2 levels has been associated with total Tau and phosphorated Tau (p-Tau, Thr181) protein levels (Suarez-Calvet and others 2016). Furthermore, in TauPS2APP mice, an AD model expressing human mutated forms of amyloid precursor protein (APP), presenilin 2 (PS2), and Tau, deficiencies in Trem2 lead to an increased and more rapid accumulation and spread of Tau, a process that is notably influenced by amyloid pathology (Lee and others 2021). Such interaction between Trem2 and amyloid pathology suggests a complex interplay in the progression of AD. Understanding the mechanisms underlying Trem2’s influence on tau and amyloid pathology could provide valuable insights into potential therapeutic targets for AD.
The crosstalk between Trem2 and mTOR signaling
Trem2 also promotes microglia survival by activating the PI3K/Akt/mTOR signaling pathway, regulating microglia autophagy, and sustaining their cellular energy and biosynthetic metabolism (Ulland and others 2017). Intriguingly, our research has revealed that activation of mTOR within microglia leads to an increase in Trem2 messenger RNA and protein expression, thereby enhancing the removal of Aβ plaques, a key factor in AD pathologies (Shi and others 2022). Furthermore, our study highlights the role of Trem2 in mediating microglia anti-inflammatory responses, lysosomal functions, and phagocytic effects in a disease state of microglia during AD pathology, as summarized in Figure 4. Addressing the lysosomal dysfunction in microglia, through mechanisms dependent on or independent of mTOR, can ameliorate issues related to lysosomal, lipid, and activation states that arise due to TREM2 mutations (Carosi and Sargeant 2019). Therefore, the interplay between Trem2 and mTOR signaling in microglia offers a promising avenue for addressing the complex pathologies associated with AD, especially those related to lysosomal and metabolic dysfunction.
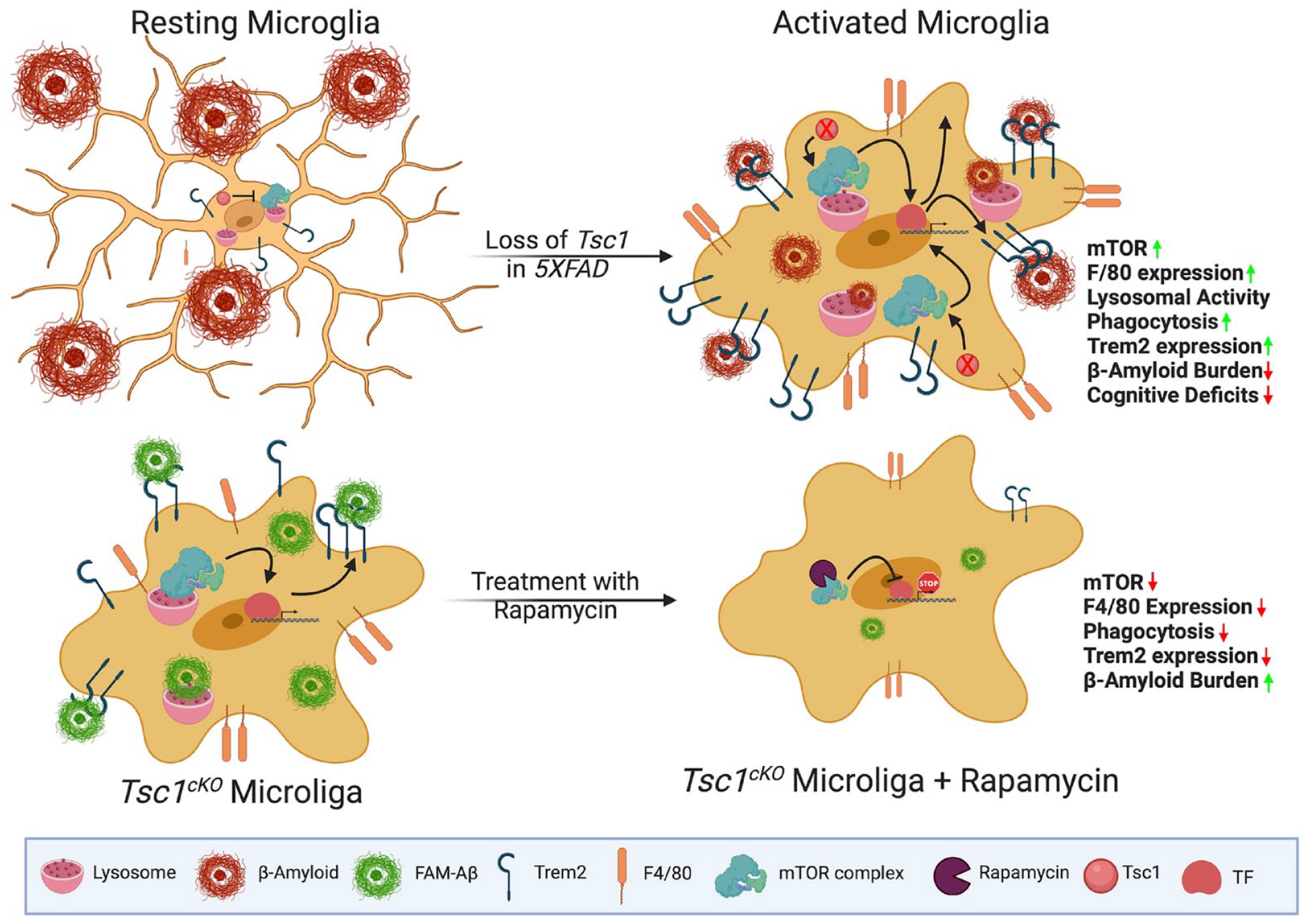
Mammalian target of rapamycin (mTOR) regulates the expression of Trem2 in microglia. Loss of Tsc1 triggers mTOR activation, which in turn stimulates microglial activation (enhanced F4/80 expression) and elevates Trem2 expression at both protein and messenger RNA levels, facilitated by specific transcription factors (TFs). This cascade enhances microglial functions, including anti-inflammatory responses, lysosomal activity, and the phagocytosis of amyloid β (Aβ) plaques, contributing to a potential reduction in Alzheimer disease (AD) pathology. Conversely, inhibiting mTOR in microglia with rapamycin leads to a decrease in Trem2 expression, resulting in lysosomal dysfunction and diminished uptake of Aβ conjugates. This reduction in microglial functionality exacerbates AD pathology and accelerates cognitive decline. This figure was created with the BioRender software.
Recent epidemiological studies have provided further insights into the prevalence and impact of TREM2 variants. While variants like R47H are rare, their presence significantly increases the risk of AD, comparable to that of carrying one copy of the APOE ε4 allele (Krasemann and others 2017). While the frequency of these variants in the general population is low, their impact on individuals carrying them is substantial, offering novel perspectives on the pathogenesis of AD and potential targets for therapeutic interventions. As genetic and functional studies continue, the implications of TREM2 variants in AD and their potential in personalized medicine will become increasingly clear.
Role of Trem2 in Other Neurodegenerative Conditions
While the role of Trem2 in AD has been extensively studied, its implications in other neurodegenerative diseases like PD and others are gaining increasing attention (Gu and others 2023; Huang and others 2024). Trem2 has been found to play a significant role in the pathogenesis and progression of various neurodegenerative conditions, due to its unique regulatory role in maintaining microglia metabolism (Gratuze and others 2020). PD is characterized by the loss of dopaminergic neurons in the substantia nigra and the accumulation of α-synuclein aggregates (Calabresi and others 2023). Recent studies have begun to unravel the role of Trem2 in PD (Li and Zhang 2021). In patients with PD, TREM2 expression is up-regulated in microglia around the α-synuclein deposits (Guo and others 2019; Zhang and others 2018). This suggests a role for Trem2 in the microglial response to neuronal damage and protein aggregation. Trem2 is believed to modulate the inflammatory response in PD. Its activation may help in shifting microglia to a neuroprotective phenotype, potentially reducing neuroinflammation and slowing disease progression (Zhang and others 2018). Trem2 might also influence the clearance of α-synuclein aggregates, a critical aspect of PD pathology. Efficient phagocytosis of these aggregates could mitigate their toxic effects on neurons. Furthermore, there is evidence that overexpression of Trem2 attenuates neuroinflammation and offers protection to dopaminergic neurons in experimental models of PD (Ren and others 2018). This finding reinforces the idea that Trem2 plays a protective role in the context of PD, particularly in terms of modulating microglial activation and the resultant inflammatory response.
Trem2 is also an important regulator in other neurodegenerative diseases. In ALS, a disease characterized by degenerating motor neurons, Trem2 expression is significantly altered in microglia (Jerico and others 2023; Xie and others 2022b). These studies have consistently shown that Trem2 could influence the survival of motor neurons by modulating microglial responses to neuronal injury and then regulating neuroinflammatory processes. Trem2 is also implicated in conditions such as FTD and Nasu-Hakola disease (NHD). NHD, in particular, is characterized by symptoms like demyelination, early-onset dementia, and bone cyst lipomas. This disease is known to be linked with specific mutations in the ectodomains of Trem2, namely, Y38C, W50C, T66M, and V126G (Guerreiro and others 2013a; Guerreiro and others 2013b; Kleinberger and others 2017; Le Ber and others 2014). Given the overlap in microglial activation patterns between FTD and other neurodegenerative diseases, Trem2 might influence disease progression through similar mechanisms of modulating microglial activation and inflammatory responses (Cady and others 2014). In MS, which is characterized by demyelination and neuroinflammation, Trem2’s role is rather complex. While it may contribute to the inflammatory response in the early stages, Trem2 also appears to be involved in the resolution of inflammation and tissue repair at later stages (Zhou and others 2019).
Overall, the role of Trem2 in many neurodegenerative disorders is increasingly recognized as crucial due to its ability to alter microglial immune activity and response. This includes modulating the balance between proinflammatory and anti-inflammatory responses in the brain, as well as altering the removal of damaged neurons and abnormal protein aggregates. Such diverse functions place Trem2 at the forefront of neuroprotective mechanisms, making it a key target for potential therapeutic strategies (George 2023). By reprogramming microglial metabolism, regulation of Trem2 activity could significantly impact the progression of various neurodegenerative diseases like AD, PD, ALS, FTD, and MS. This opens new possibilities for developing treatments aimed at slowing the advance of these conditions and reducing the harm to neurons and accordingly the neuronal circuits.
Therapeutic Implications and Challenges
Trem2 as a Therapeutic Target in AD
The microglia-Trem2 axis has emerged as a pivotal target in the treatment of neurodegenerative diseases, with Trem2 playing a crucial role in regulating numerous microglial functions. This includes phagocytosis, inflammation, and neuroprotection. Modulating the interactions between microglia and Trem2 offers potential therapeutic strategies for conditions like AD, PD, and other neurodegenerative disorders (George 2023). Preclinical studies have strongly suggested a protective role of microglial Trem2 in relations to the Aβ-associated AD pathogenesis (Hou and others 2022) as well as the Aβ-induced Tau pathology (Leyns and others 2019). Developing agonistic antibodies that enhance Trem2 activity could boost microglial phagocytosis and anti-inflammatory responses (Schlepckow and others 2023), particularly beneficial in diseases characterized by the accumulation of toxic proteins, such as Aβ in AD. The Trem2 antibody 4D9 was the first agonistic antibody developed against the stalk region of Trem2 and reduces its proteolytic shedding, consequently activating downstream SYK signaling, which enhances the survival of murine macrophages (Schlepckow and others 2020). In line with this finding, other studies have shown that two monoclonal antibodies, AL002c and AL002a (Price and others 2020; Wang and others 2020), tailored for human and mouse Trem2, respectively, have been confirmed to ameliorate Aβ pathology in vivo, supporting the therapeutic potential of targeting Trem2. Similarly, another monoclonal antibody, CGX101, which binds to the extracellular domain of human and murine Trem2, has been shown to enhance cellular proliferation, promote uptake of Aβ or apoptotic neurons, and activate SYK and AKT signaling pathways. These effects of CGX101 suggest its potential to improve the cognitive functions in patients with AD (Fassler and others 2021). In summary, the development of antibodies targeting Trem2 represents a promising therapeutic approach in AD, which highlights the critical role of Trem2 in AD pathology and offers a novel avenue for treatment.
Additionally, gene therapy techniques, such as viral vector–mediated gene delivery, could be used to increase Trem2 expression in brain microglia, potentially restoring the functional deficits observed in AD or other neurodegenerative diseases. A recent study (Wang and others 2022) introduced a microglia-targeted gene delivery system, PHSA@PF/pTREM2, which aims to up-regulate Trem2 in microglia. This system has proved to cross the blood-brain barrier, target microglia efficiently, and release the Trem2-encoding plasmid in the acidic environment of endo-lysosomes. This system enhanced Trem2 expression in microglia, which shifted their polarization toward the M2 phenotype. This further promoted Aβ clearance and inhibited neuroinflammation, potentially halting or reversing AD progression in APP/PS1 mouse models. Alternatively, Wu et al. (Wu and others 2022) used an adeno-associated virus–mediated Trem2 overexpression in the hippocampus in a high-fat diet mouse model. This Trem2 overexpression led to an increase in synaptic proteins and normalized dendritic complexity and spine density in the hippocampus by suppressing NF-κB pathway and neuroinflammation, which could be potentially useful in an AD model. Not only is the affinity of the antibody itself to Trem2 of importance, but the transport system delivering the antibody is also at the center of pharmaceutical consideration. Recently, a novel therapeutic approach has been developed for AD using a Trem2-activating antibody combined with the transferrin receptor binding site for enhanced brain delivery (van Lengerich and others 2023). This therapy, named ATV:TREM2, showed improved brain tissue distribution and increased Trem2-dependent downstream signaling as compared to a standard anti-Trem2 antibody delivery. This could represent a promising new approach to improve microglial functionality and potentially a treatment for patients with AD. All abovementioned therapeutic approaches targeting Trem2 are demonstrated in Figure 5.
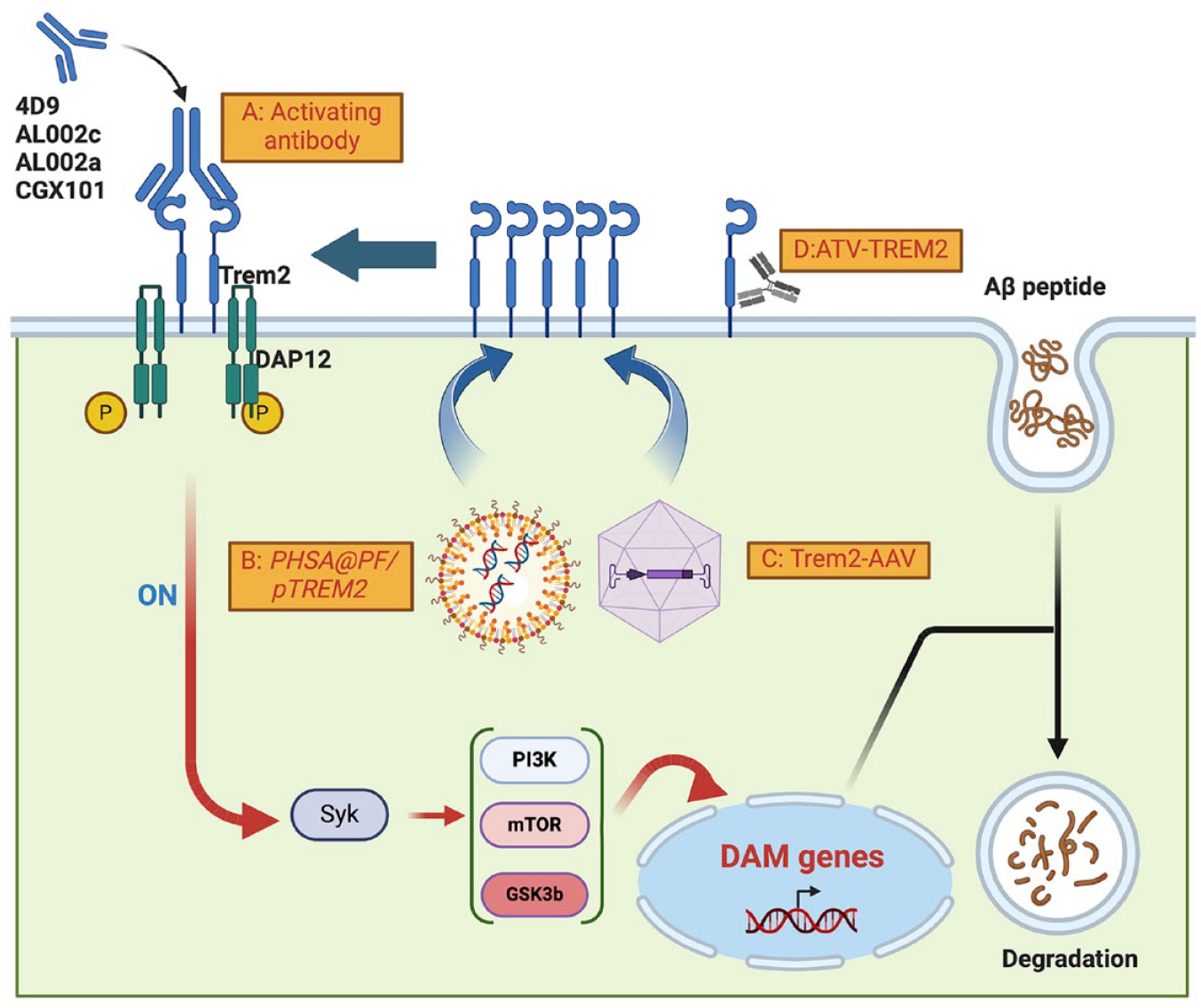
Targeting Trem2 for therapeutic intervention in Alzheimer disease (AD) and neurological disorders. (A) Agonistic antibodies: antibodies such as 4D9, AL002c, AL002a, and CGX101 specifically bind to Trem2 on the microglial cell membrane. This interaction boosts microglial phagocytosis and anti-inflammatory activities, contributing to the reduction of amyloid β (Aβ) pathology and enhancement of cognitive functions in AD models. (B) Viral PHSA@PF/pTREM2 gene delivery: systems like PHSA@PF/pTREM2 use viral vectors to elevate Trem2 expression on the microglial membrane. This strategy aims to enhance Aβ clearance, reduce neuroinflammation, and improve synaptic integrity, addressing key aspects of AD pathology. (C) Adeno-associated virus (AAV)–mediated Trem2 overexpression: similar to (B), this approach employs AAV vectors to increase Trem2 expression in microglia. The overexpression of Trem2 is designed to promote the removal of Aβ, mitigate inflammatory responses, and support the restoration of neuronal connections. (D) Enhanced brain delivery via TREM2-activating antibodies: a novel therapeutic strategy combines TREM2-activating antibodies with a transferrin receptor (TfR) binding site, significantly improving the brain distribution and effectiveness of TREM2 protein. This method shows great promise in enhancing microglial functionality and offering a new treatment pathway for patients with AD. This figure was created using the BioRender software.
A microglial cell therapy would be a potential option in conjunction with the same concept of boosting Trem2 expression in neurological disorders that are either TREM2 mutant or deficient. In AD mouse models, transplanting or substituting wild-type circulation-derived myeloid cells (CDMCs) for Trem2 mutant or Trem2 KO microglia resulted in the restoration of microglia activation in vivo, a decrease in Aβ plaque formation, and the induction of plaque compaction (Yoo and others 2023). This strategy led to the restoration of the DAM microglia transcriptional program and the downstream SYK signaling pathway, as well as an increase in microglial surface receptors such CD11c and Clec7a. These findings imply that effective cell therapy could be a promising treatment for AD. Along the same line, indirectly modulating Trem2 expression could offer a different avenue for developing a new targeting therapy for AD. The traditional study had shown that Trem2 regulates PI3K/Akt/mTOR signaling pathways by directly interacting with DAP12/SYK molecules (Ulland and others 2017; Zhou and others 2018). However, we have discovered that the alteration in the mTOR signaling pathway significantly changes the expression levels of Trem2 in microglia during AD pathogenesis (Shi and others 2022). This change significantly reduced Aβ plaque buildup and improved cognitive performance in the 5xFAD AD mouse model. On the other hand, rapamycin treatment on 5xFAD mice, which inhibits mTOR, significantly increased AD progression, marked by less responsive microglia and increased Aβ plaque formation. These findings align with recent suggestions (Carosi and Sargeant 2019) that rapamycin could adversely affect AD treatment due to its impact on mTOR signaling. Administering rapamycin to patients with dementia might exacerbate microglial dysfunction and promote further accumulation of Aβ plaques, intensifying AD-related pathologies. Also, small molecules that can modulate Trem2 activity offer a more traditional pharmacological approach. These could be designed to either enhance or inhibit Trem2 signaling, depending on the disease context. It was recently reported that inhibitors of MEK1/2 kinases significantly increase cell surface Trem2 protein expression, suggesting a new or indirect pathway for Trem2 regulation (Schapansky and others 2021). However, direct binding of small molecules to enhance or inhibit Trem2 expression remains to be identified.
Thus, research efforts focused on the microglia-Trem2 relationship provides a promising direction for treating neurodegenerative diseases. Diverse therapeutic methods, including agonists, antagonists, gene therapies, cell therapy, and small molecules, are being developed to influence this pathway. However, given the varied responses of microglia and the diverse nature of neurodegenerative diseases, personalized and stage-specific approaches are needed. A better mechanistic knowledge about the microglia-Trem2 biology promises to significantly advance treatment options for neurological disorders that directly interface with microglia.
Challenges and Future Research Directions
The field of targeting the microglia-Trem2 axis in neurodegenerative diseases offers therapeutic promises but is also fraught with challenges that require careful navigation. One of the primary challenges is the complexity of microglial responses at different disease stages (Hickman and others 2018; Song and Colonna 2018). Microglia exhibit a spectrum of activation states and responses, and their role in neurodegenerative diseases is not entirely linear. Overactivation can lead to chronic inflammation and exacerbate neuronal damage, while underactivation can result in insufficient clearance of debris and misfolded proteins. Future research should focus on delineating the precise activation states of microglia in various neurodegenerative conditions and understanding how Trem2 modulation affects and alters these states. Another challenge is the heterogeneity of neurodegenerative diseases. Each condition, from AD to PD, has unique pathological features and molecular mechanisms. This diversity means that a one-size-fits-all approach to modulating the microglia-Trem2 axis is unlikely to be therapeutically effective. The potential side effects of modulating Trem2 activity also pose challenges. While enhancing Trem2 function might be beneficial in certain contexts, it could potentially lead to unwanted side effects, such as excessive immune responses or interference with normal brain homeostasis. Microglia are not the only cells that express Trem2; targeting of this receptor outside the brain will need to be assessed to make sure there are no unwanted side effects. Finally, the development of biomarkers for microglial activation and Trem2 function, particularly in the peripheral systems, could significantly advance the field. Such biomarkers would enable the monitoring of disease progression and treatment response, facilitating the development of more effective therapies. Future research should focus on identifying and validating biomarkers that reflect the real-time state of the microglia-Trem2 axis in vivo in both healthy and disease states.
In conclusion, while targeting the microglia-Trem2 axis in neurodegenerative diseases holds great promise, it is a field with many challenges. Addressing these challenges through focused research will be crucial for developing effective and safe therapies. Future research directions should delve into providing a deeper mechanistic understanding of microglial biology, disease- and stage-specific strategies, development of relevant biomarkers, and personalized cell or gene therapy approaches, which will bridge the translational gap and thus help patients with various neurodegenerative diseases.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Zachry Foundation Endowment for advancing neuroscience research, the Doran Family Foundation and the University of Texas Health San Antonio (M.A.B.). The authors received no other financial support for the research, authorship, and/or publication of this article.