
Abstract
Unraveling the neuronal mechanisms of fear learning might allow neuroscientists to make links between a learned behavior and the underlying plasticity at specific synaptic connections. In fear learning, an innocuous sensory event such as a tone (called the conditioned stimulus, CS) acquires an emotional value when paired with an aversive outcome (unconditioned stimulus, US). Here, we review earlier studies that have shown that synaptic plasticity at thalamic and cortical afferents to the lateral amygdala (LA) is critical for the formation of auditory-cued fear memories. Despite the early progress, it has remained unclear whether there are separate synaptic inputs that carry US information to the LA to act as a teaching signal for plasticity at CS-coding synapses. Recent findings have begun to fill this gap by showing, first, that thalamic and cortical auditory afferents can also carry US information; second, that the release of neuromodulators contributes to US-driven teaching signals; and third, that synaptic plasticity additionally happens at connections up- and downstream of the LA. Together, a picture emerges in which coordinated synaptic plasticity in serial and parallel circuits enables the formation of a finely regulated fear memory.
Keywords
Introduction
Animals must predict threats in their environment using sensory cues and react with appropriate defensive behaviors to ensure survival. For this reason, threat-predicting neuronal networks have evolved in animals (Feinberg and Mallatt 2017; Seymour 2019). The amygdala has long been recognized as a brain area that integrates and processes information about sensory cues and that is involved in the execution of defensive or approach behaviors, depending on the valence of the perceived cues (Janak and Tye 2015; Paton and others 2006). In aversively motivated learning, the internal emotional state caused by the detection of a threat is referred to as “fear” (LeDoux 2014). In behavioral experiments, defensive behaviors or other motor outputs, such as autonomic responses, can be observed as a proxy for the internal fear state of an animal (Fanselow 1994; LeDoux and others 1988).
Fear conditioning paradigms have been used for several decades in rodents to investigate the brain areas and synaptic connections that underlie aversively motivated learning (LeDoux 2000; Maren 2001; Tovote and others 2015). During fear conditioning, an initially neutral sensory cue, the conditioned stimulus (CS), comes to predict an aversive outcome, the unconditioned stimulus (US), after being paired in a predictable manner with the latter. Following fear learning, the CS itself elicits a defensive behavior, such as freezing (Fanselow 2018; Fig. 1). Fear memories are maintained over long times, but it is important to note that the brain sites of memory storage change with time (DeNardo and others 2019; Do-Monte and others 2015). In humans, strong traumatic events can cause dysregulated fear memories and posttraumatic stress disorder (Ressler and others 2022; Yehuda and others 2015). Therefore, an improved understanding of the molecular, cellular, and circuit-level mechanisms of plasticity that underlie fear learning holds the promise to enable the development of advanced therapies of anxiety disorders.
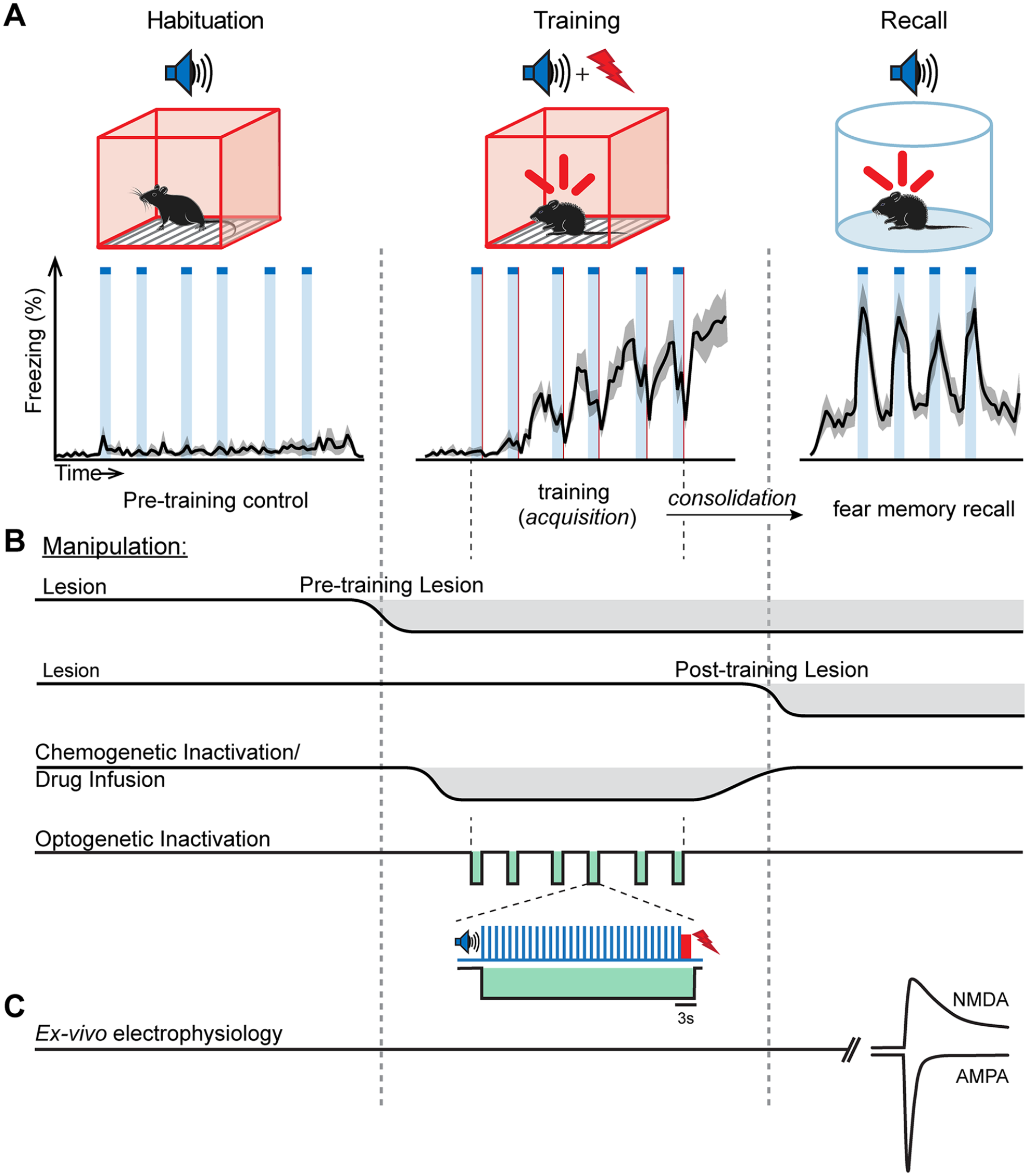
Behavioral paradigm and circuit manipulations in fear learning. (A) Top: scheme of the basic auditory-cued fear conditioning protocol used in many studies. Bottom: average freezing trace of 10 mice undergoing the fear learning protocol, binned over 10-s intervals (S. Palchaudhuri, D. Osypenko, O. Kochubey and R. Schneggenburger, unpublished). (B) Time line of in vivo manipulations of neural circuits at different phases of the fear learning protocol. (C) Illustration of the ex vivo approach to measure properties of synaptic transmission at defined synapses following fear learning. As an example, measurement of the AMPA/NMDA ratio at a glutamatergic synapse recorded at negative and positive membrane potentials is shown.
The release of neurotransmitters at synapses is a key biological process that enables dynamic communication between neurons (Jessell and Kandel 1993; Südhof 2013). For the present review, excitatory connections that allow the communication between distant brain areas are of special relevance. Since its discovery, long-term potentiation (LTP) at excitatory synapses has been discussed as a cellular correlate underlying learning and memory (Bliss and Collingridge 1993; Nicoll 2017; Stevens 1998). In this respect, fear learning and the amygdala circuits have been an important model to trace the relationship between cellular plasticity at specific synaptic connections, and a learned behavior (Sigurdsson and others 2007).
Here, we review key studies that have linked synaptic plasticity at specific input synapses to the amygdala, with behavioral fear learning (for earlier reviews, see Pape and Pare 2010; Sigurdsson and others 2007). We concentrate on studies that described LTP at long-range synaptic inputs to the lateral nucleus of the amygdala, an important input nucleus of the amygdala complex (LeDoux and others 1990). We then complement these findings with results from more recent studies, which employed newer methods of in vivo imaging of neuronal activity and optogenetic methods to dissect the circuit mechanisms of fear learning. We do not discuss the role of local inhibitory circuits in fear learning, but excellent recent reviews exist on this topic (Cummings and others 2021; Krabbe and others 2018). We apologize to those authors whose papers we could not discuss because of space limitations.
Fear Learning: Behavioral Paradigm and Circuit Manipulations
Mice have become an important model species for studying the circuit mechanisms underlying fear learning, because of their genetic accessibility, allowing the generation of Cre lines to target specific neuronal populations (e.g., Madisen and others 2010; Matho and others 2021; Taniguchi and others 2011; Tsien and others 1996). Also, optogenetic experiments can readily be performed in mice, and because of the genetic accessibility of this model species, these manipulations can be endowed with cell-type specificity (Chow and others 2012; Sparta and others 2012; Tye and Deisseroth 2012). Moreover, techniques to record or image the in vivo activity of neuronal populations in freely moving mice have been devised and optimized in recent years (Anikeeva and others 2012; Ghosh and others 2011; Jun and others 2017).
A typical auditory-cued fear learning paradigm, as used in rodents, is illustrated in Figure 1. As a CS, brief beeps of tone stimuli repeated for several tens of seconds are typically used (Rogan and others 1997); naive mice do not freeze in response to such auditory stimuli. On the training day, the CS is typically paired several times with a footshock (Fig. 1A), although a single pairing is sufficient to drive fear learning (Krabbe and others 2019). During the training session, mice demonstrate a gradual increase in freezing that is not yet time locked to the CS (Fig. 1A, middle). One day later, when the animal is placed in a different context, a swift increase in freezing time-locked to the CS is now apparent. In addition, there is a lower level of time-invariant freezing, which is likely caused by unavoidable contextual cues (Fig. 1A, right). The auditory-driven conditioned response demonstrates that the CS representation has gained access to a defensive behavior after a phase of fear memory consolidation (Fanselow 2018).
Experimental loss-of-function manipulations have been performed to investigate brain areas and synaptic circuits relevant for fear learning. These include lesions of specific brain areas, drug infusion experiments and pharmacogenetic inactivation, as well as, more recently, optogenetic silencing (Fig. 1B). Lesions can either be made before the training session, or posttraining, depending on whether the role of a brain area is tested in the acquisition or in the consolidation of a fear memory (Fig. 1B; see Rodrigues and others 2004 for an early review). However, lesions cause permanent removal of a given brain area, and given the redundancy of synaptic connections in the brain (e.g., Bota and others 2015), there might be compensation by other brain areas. In another approach, drugs that block signaling pathways involved in plasticity can be infused locally, an approach that can test a particular molecular hypothesis of learning (Rodrigues and others 2004). In yet another approach called pharmacogenetic silencing, neurons in a brain area can be transiently inactivated, also in a cell type–specific manner (Armbruster and others 2007). Finally, optogenetic silencing of neurons has an improved timing precision—in principle, down to the millisecond range (Chow and others 2010; Gradinaru and others 2010; see Wiegert and others 2017 for a review). Optogenetic silencing can, for example, be used to suppress neuronal activity during the entire CS-US pairing protocol (Barsy and others 2020; Fig. 1B, bottom), or it can be restricted to selectively cover the footshock stimulation (Johansen and others 2014; Namburi and others 2015; Uematsu and others 2017). In chemogenetic and optogenetic experiments, the desired reduction of neuronal action potential firing activity in a targeted neuronal population should best be validated in vivo.
Studies of LTP at hippocampal synapses had shown that a prevalent mechanism of LTP is the insertion of AMPA-type glutamate receptors into the postsynaptic membrane (Choquet and Hosy 2020; Malenka and Nicoll 1999; Malinow and Malenka 2002; Nicoll 2003). Correspondingly, measurements of the AMPA/NMDA ratio at a given synaptic connection can be used to infer the plasticity state of a synapse (Bellone and Lüscher 2006; Clem and Huganir 2010; Rothwell and others 2015; Shan and others 2014) (Fig. 2A). Because it is difficult to measure synaptic transmission with high resolution in vivo, such measurements have been performed “ex-vivo”, to correlate signs of plasticity at specific synapses, with previous fear learning in the living animal (Fig. 1C). The studies were initially performed with electrical stimulation of fiber pathways. More recently, they are often combined with optogenetically assisted circuit mapping, which enables the optical stimulation of a set of synapses arising from an upstream brain area (Kim and Cho 2017; Petreanu and others 2007; Rich and others 2019; Yu and others 2017).
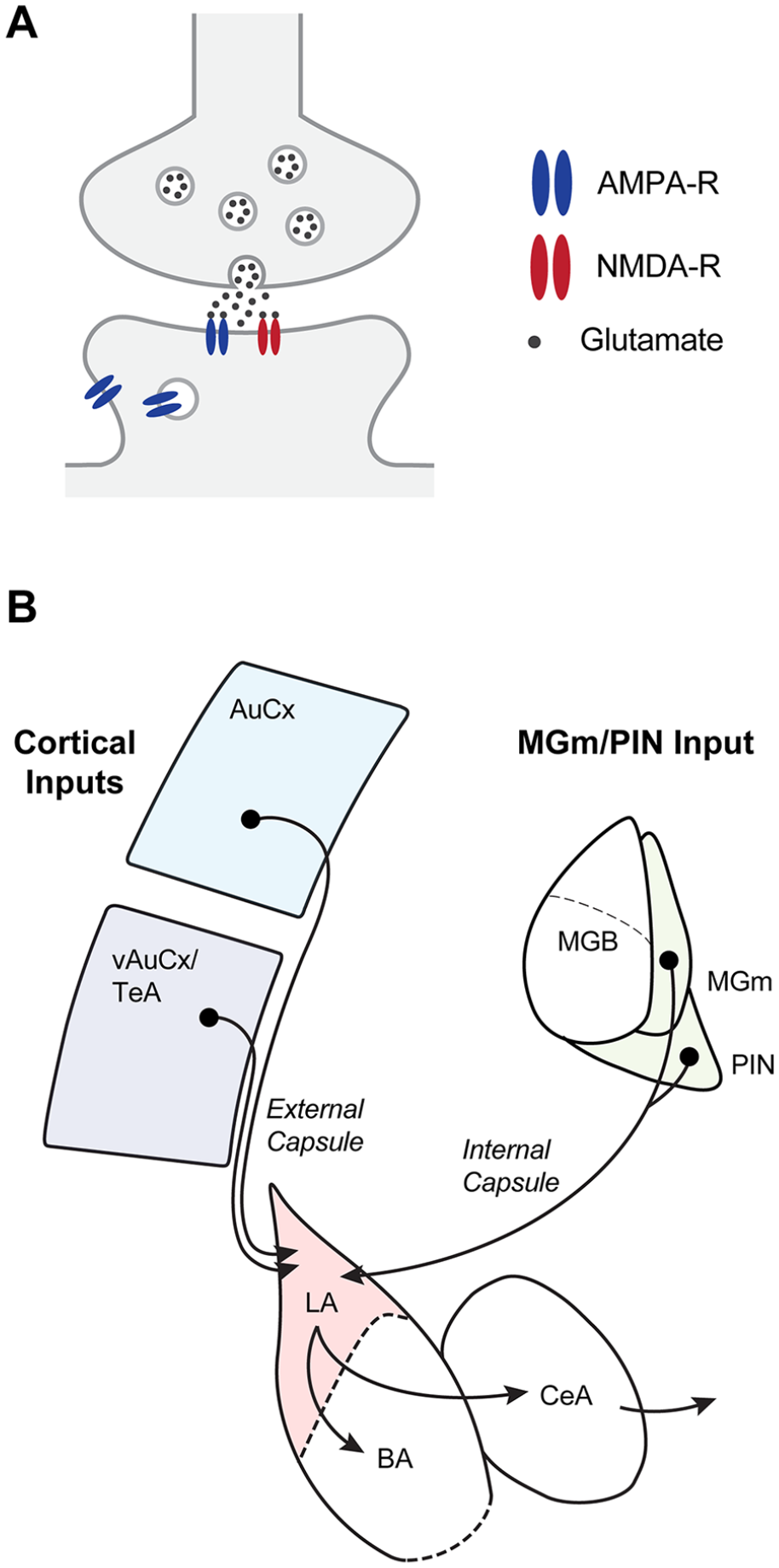
Some afferent synapses to the amygdalar complex. (A) Information enters the amygdala via long-range glutamatergic connections. A glutamatergic synapse with AMPA and NMDA receptors is illustrated. (B) Scheme of the most important afferents and efferents as relevant for the studies reviewed here. The inputs to the lateral amygdala (LA) most relevant for auditory-cued fear learning are from various cortical areas, such as the auditory cortex (AuCx) as well as the auditory association areas of the ventral auditory cortex and the temporal association cortex (abbreviated here together as the vAuCx/TeA). Likewise, thalamic inputs from the medial part of the medial geniculate nucleus (MGm) and the posterior intralaminar nucleus (PIN; referred to here as MGm/PIN) have been described. BA = basal amygdala; CeA = central amygdala; MGB = medial geniculate body (the main part of the auditory thalamus).
Role of Amygdalar Substructures in Fear Learning
Early lesion studies showed a role for the amygdala in fear learning (see LeDoux 2000; Maren 2001 for reviews). Lesions in the lateral amygdala (LA) and central amygdala (CeA) were especially effective in suppressing the acquisition of auditory-cued fear memories (Campeau and Davis 1995a; LeDoux and others 1990; Nader and others 2001; see also Goosens and Maren 2001).
The amygdala is a highly complex brain area with many subdivisions that have different functional roles and different embryologic origins (Hirata and others 2009; Sah and others 2003; Swanson and Petrovich 1998). For the purpose of this review, it is useful to mainly consider the lateral and basal nuclei (LA and BA) and the central nucleus (CeA) of the amygdala (Fig. 2B). The CeA is composed mainly of GABAergic local and projecting neurons (Ciocchi and others 2010; Haubensak and others 2010; Swanson and Petrovich 1998). The CeA makes outputs to hypothalamic and midbrain areas to coordinate and initiate defensive behavioral responses, including freezing and autonomic responses (LeDoux and others 1988; Tovote and others 2016). Thus, the CeA can be regarded as a striatal-like circuit and as the output structure of the amygdala, at least concerning learned fear responses.
In contrast to the CeA, the LA and BA are cortical-like structures containing a majority of excitatory principal neurons and local inhibitory interneurons (Sah and others 2003; Swanson and Petrovich 1998). As we review below, the LA receives sensory inputs from cortical and thalamic areas, whereas the BA is connected with limbic areas such as the prefrontal cortex and hippocampus (Hintiryan and others 2021). Furthermore, a recent study demonstrated differential gene expression between neurons in the LA and the BA of mice (O’Leary and others 2020). For these reasons, we refer to the LA and BA separately in this review, whenever the original work allows such a separation. In this terminology, the BA corresponds roughly to the BLA and BLP of mouse brain atlases (for “basolateral amygdaloid nucleus, anterior and posterior part,” respectively; Allen Institute for Brain Science 2011; Paxinos and Franklin 2019).
Afferents to the Amygdala and Their Role in Fear Learning
In parallel to the lesion studies that identified important roles of the LA and CeA in fear learning, early neuroanatomic and functional studies revealed that the LA is an important station for sensory inputs to the amygdalar complex (Campeau and Davis 1995b; LeDoux 2000). The LA is innervated by various cortical areas, such as the auditory cortex (AuCx) and the ventral auditory cortex and temporal association cortex (vAuCx/TeA), as well as by thalamic areas, such as the medial geniculate nucleus/posterior intralaminar nucleus (MGm/PIN; Doron and Ledoux 2000; Hintiryan and others 2021; LeDoux and others 1991; McDonald 1998; Morikawa and others 2021; Fig. 2B). Thus, one could say that the LA is innervated by “auditory” cortical and thalamic areas. However, this view is likely an oversimplification because thalamic areas such as the MGm/PIN code not only for auditory modalities but also for somatosensory/aversive events (Barsy and others 2020; Bordi and LeDoux 1994; Taylor and others 2021; see Weinberger 2011 for a review of earlier studies). Indeed, MGm/PIN are not part of the tonotopically organized ventral medial geniculate body (MGB) that sends information to the primary AuCx, but rather are so-called higher-order or nonlemniscal thalamic areas involved in a range of functions (Lee 2015; Lee and Sherman 2011). Similarly, the vAuCx/TeA, which sends a strong connection to the LA, has recently been shown to integrate information about aversive somatosensory stimulation and to code for the negative emotional qualities of a tone during fear learning (Dalmay and others 2019). Thus, the auditory afferents likely already contain aversive somatosensory information, a finding that is relevant for models of fear learning (see below). In addition, the LA is innervated by other brain areas that have been less well studied in the context of fear learning (Hintiryan and others 2021; Morikawa and others 2021).
Interestingly, early lesion studies have already addressed the question of whether the thalamic or cortical afferents (or both) are involved in auditory-cued fear learning. This was done by selectively lesioning the MGm/PIN (the nonlemniscal auditory thalamus), or the MGB, or else the auditory cortical areas of rats (Campeau and Davis 1995b; Romanski and LeDoux 1992). An implicit assumption of these studies was that the main effect of the lesions would be to interrupt the flow of sensory information to the LA, but that these thalamic and cortical structures would not, by themselves, have a role in fear learning. In an asymmetrical lesion approach in which the entire auditory thalamus on one brain side was damaged, Romanski and LeDoux (1992) found that lesioning, on the other brain side, the MGm/PIN or the AuCx alone did not affect fear learning. However, combined lesions of both areas caused a strong impairment of auditory-cued fear learning. Similarly, Campeau and Davis (1995b) reported that bilateral lesions of the entire auditory thalamus strongly impair fear learning as measured by fear-potentiated startle, whereas spatially confined lesions of the MGm/PIN left fear learning largely unchanged. These studies showed that the primary and nonlemniscal auditory thalamus are together strictly necessary for auditory-cued fear learning. On the other hand, bilateral lesions of the MGm/PIN alone was apparently compensated by the flow of sensory information from the MGB to auditory cortical areas, and from there to the LA (Fig. 2B). Thus, at least in basic forms of auditory-cued fear learning, there is a certain degree of “equipotentiality” of the direct thalamo-LA pathway, and of the indirect thalamo-cortical-amygdala pathways (Romanski and LeDoux 1992).
When mice or rats are trained for longer times with two auditory CSs, one that is paired with a footshock and one that is not (CS+ and CS–, respectively), they learn to discriminate between the tones, which can differ, for example, by their sound frequencies (Antunes and Moita 2010; Dalmay and others 2019). Early studies based on auditory conditioning of heartbeat rate in rabbits showed that bilateral lesions of the AuCx suppress this discriminative fear learning (Jarrell and others 1987). A more recent study with transient inactivation of the AuCx by the GABAA agonist muscimol in rats demonstrated that AuCx activity is necessary for the acquisition and retrieval of discriminative fear learning (Wigestrand and others 2017). Furthermore, by using infusion of a PKM-zeta inhibitor, this study reported that plasticity in AuCx is necessary for discriminative fear learning. These studies support the view that the pathway from the MGB via the auditory cortical areas and finally to the LA is necessary for discriminative fear learning. Some studies found that in addition to the AuCx, the MGm/PIN is necessary for discriminative fear learning (Antunes and Moita 2010; Jarrell and others 1987). Nevertheless, it remains possible from these lesion studies that the MGm/PIN has a role in discriminative fear learning via outputs to brain areas beyond the LA.
A recent study reinvestigated the role of the different auditory cortical areas in discriminative and basic forms of fear learning, by using optogenetic silencing and activation approaches and in vivo two-photon Ca2+ imaging in mice. The study reported that the primary AuCx is necessary when animals have to distinguish more complex sounds in fear learning, such as frequency-modulated sweeps with opposite directions (Dalmay and others 2019). On the other hand, neurons in the more ventrally located associative areas (called here vAuCx/TeA; Fig. 2B) were involved in more basic forms of fear learning with simpler tones. Moreover, the vAuCx/TeA had a higher density of LA projection neurons than the primary AuCx. In vivo two-photon Ca2+ imaging showed that on the population level, these LA projectors can distinguish CS+ and CS– stimuli, and optogenetic stimulation of their axons induced freezing in naive mice (Dalmay and others 2019). These results illustrate a gradient of functions of auditory cortical areas in different forms of fear learning, with a role in more complex forms of fear learning for the more dorsal AuCx areas.
We now review studies that found evidence for synaptic plasticity at thalamic and cortical inputs to the LA in fear learning.
Plasticity at Thalamic Inputs to the LA during Fear Learning
A series of early studies employing in vivo recordings in rats investigated the hypothesis that auditory information might flow from the MGm/PIN to the LA during fear learning, and that a process of LTP at this connection might underlie auditory-cued fear learning. These studies found an increased information transmission from the MGm/PIN to the LA during fear learning (Clugnet and LeDoux 1990; Rogan and LeDoux 1995; see also Rosenkranz and Grace 2002). Addditional studies showed that fear conditioning leads to a long-lasting increase of an auditory-evoked potential in the LA that had a fast latency (<20 ms), which reflected the transmission of auditory information from the MGm/PIN to the LA (Quirk and others 1995; Rogan and others 1997).
Further studies turned to an ex vivo approach to study signs of long-term synaptic plasticity at broadly defined input synapses to the LA (Fig. 1C). Early studies investigating the thalamic input synapse to the LA used electrical stimulation of axon fibers in the internal capsule, which can be assumed to recruit axons arriving from the thalamus, including the MGm/PIN (Humeau and others 2003; McKernan and Shinnick-Gallagher 1997; Weisskopf and LeDoux 1999). We call the input activated with this technique the “thalamo-amygdala synapse” to distinguish it from studies in which inputs from the MGm/PIN were targeted more precisely by optogenetically assisted circuit mapping (Rich and others 2019; Yu and others 2017). In a pioneering study, McKernan and Shinnick-Gallagher (1997) found an increased slope of the excitatory postsynaptic current (EPSC) amplitude versus the stimulation intensity at the thalamo-amygdala synapse, in slices from animals that had previously undergone fear learning (Fig. 3A). These experiments represent, to our knowledge, the first ex vivo evidence for a change in synaptic transmission strength observed after a learning experience.
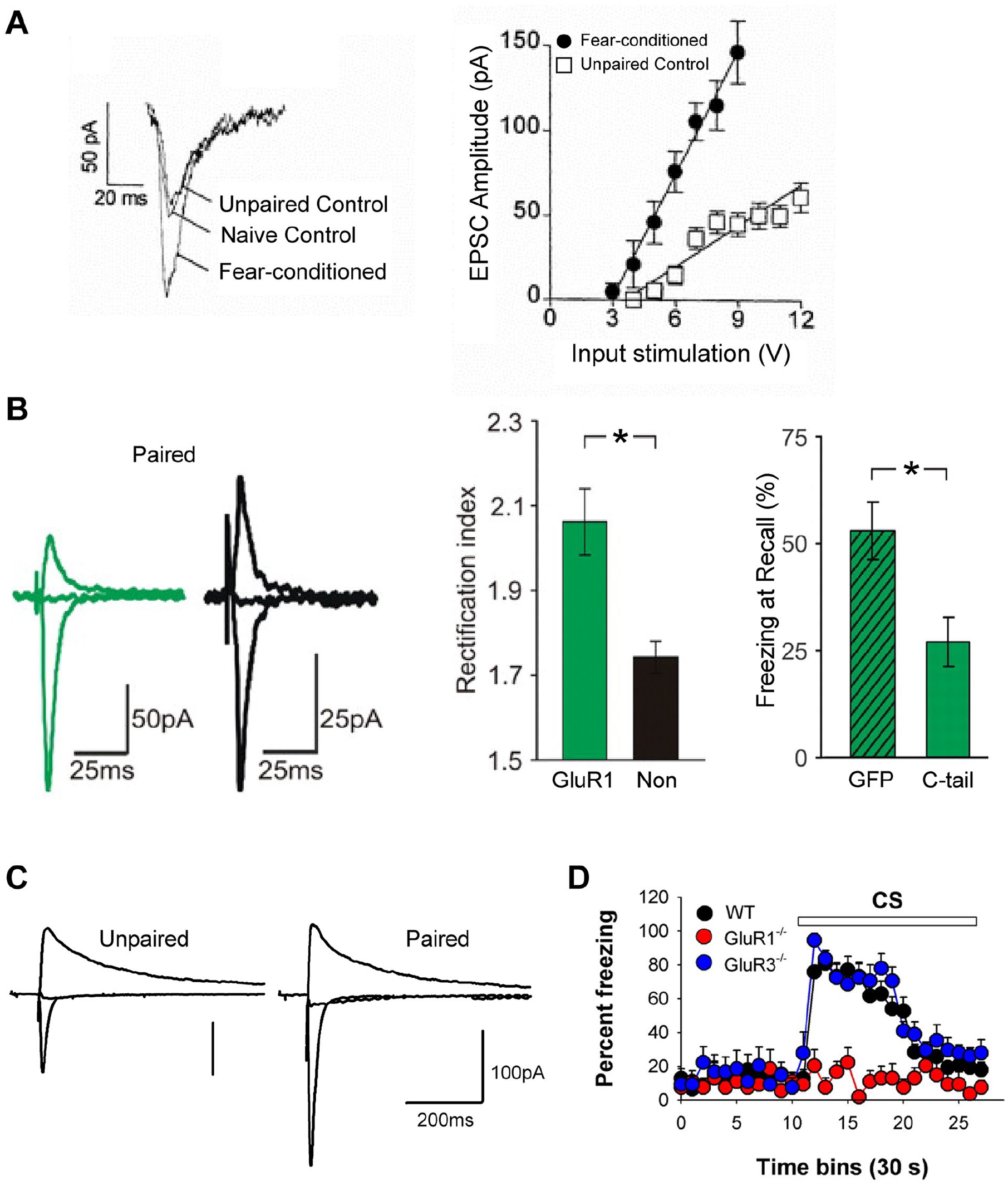
Role of plasticity at thalamic and cortical afferents to the lateral amygdala in fear learning. (A) EPSC recorded in brain slices of the lateral amygdala after electrical stimulation of the internal capsule are enhanced after fear learning, relative to unpaired or naive control animals. Reproduced with permission from McKernan and Shinnick-Gallagher (1997). (B) Fear learning drives exogenously expressed AMPA receptor GluR1 subunits into the thalamo-amygdala synapse, indicating that a postsynaptic form of long-term potentiation (LTP) occurs during fear learning. The right panel shows that overexpression of the GluR1 C-terminal domain, which blocks LTP, leads to a reduced fear memory as measured by freezing on the recall day. Reproduced with permission from Rumpel and others (2005). (C) Increased AMPA/NMDA ratio at the thalamo-amygdala synapse following fear learning, again indicating that a postsynaptic form of LTP occurs during fear learning. Reproduced with permission from Clem and Huganir (2010). (D) Strongly reduced freezing in GluR1D knockout mice during fear memory recall indicates a role for postsynaptic LTP in fear memory formation. Reproduced with permission from Humeau and others (2007). Values in A, B and D are presented as mean (SEM). *P < 0.01. CS = conditioned stimulus; EPSC = excitatory postsynaptic current; GFP = green fluorescent protein; WT = wild type.
In further experiments, the hypothesis was tested that a postsynaptic form of LTP occurs at the thalamo-amygdala synapse after fear learning. Based on the finding that the exogenously expressed GluR1 subunit of AMPA receptors is transported into synapses by LTP induction (Shi and others 2001), Rumpel and others (2005) showed that following the expression of GluR1, the rectification of AMPA-EPSCs was increased after fear learning (Fig. 3B). This strongly suggested that AMPA receptors become incorporated by LTP during fear learning at this connection. Furthermore, expression of the C-terminus of GluR1 in the LA, which blocked LTP at the thalamo-amygdala synapse, caused a ~50% suppression of auditory-cued fear memory (Rumpel and others 2005; Fig. 3B, right). This work thus demonstrated that LTP—specifically, insertion of AMPA receptors at the thalamo-amygdala synapse—contributes to the formation of an auditory-cued fear memory. Nevertheless, the blocking effect on fear memory caused by the overexpression of the GluR1 C-terminus might additionally indicate that plasticity at other input synapses to the LA, such as cortical inputs, had contributed to fear learning.
In agreement with a postsynaptic form of LTP, the AMPA/NMDA ratio at the electrically stimulated thalamo-LA synapse was increased following fear learning (Clem and Huganir 2010; Fig. 3C). In another study, neurons recorded in the BA and LA were specified according to their projection target. By again using the ex vivo approach and measuring EPSCs at the thalamo-amygdala synapse, it was found that CeA projectors had an increased AMPA/NMDA ratio after fear learning whereas, interestingly, nucleus accumbens projectors had a decreased AMPA/NMDA ratio. This indicated that neurons that might process sensory stimuli with opposite valences—here identified by their projection targets—undergo differential plasticity at their input synapses (Namburi and others 2015). Furthermore, the surface expression of GluR1- and GluR2-type AMPA receptors in the LA was increased after fear learning (Kim and others 2007; Yeh and others 2006), which provides additional evidence that postsynaptic forms of plasticity take place in the LA during fear learning.
A very interesting finding was made when knockout (KO) mice for the AMPA receptor subunit GluR1 were investigated for plasticity deficits at amygdalar synapses and behavioral deficits in fear learning. Previous work found that genetic deletion of the GluR1 leads to reduced extrasynaptic AMPA receptors, to the disappearance of LTP in hippocampal synapses (Zamanillo and others 1999), and to a spatial working memory deficit (Reisel and others 2002; Schmitt and others 2005). Investigating the GluR1 KO mice in the amygdala revealed a measurable reduction of the AMPA component to the EPSC, as well as an absence of LTP at the electrically stimulated thalamic and cortical input synapses onto LA —and separately measured— BA neurons (Humeau and others 2007). Importantly, auditory-cued fear learning was abolished in the GluR1 KO mice, with strongly reduced freezing levels on the training day and during auditory-cued fear memory recall (Humeau and others 2007; Fig. 3D). An independent study confirmed the behavioral findings and demonstrated that nociceptive responses were unchanged in GluR1 KO mice, thus excluding the possibility that the learning deficits were caused by a reduced transmission of nociceptive information (Feyder and others 2007). The studies with the GluR1 KO mouse thus showed that AMPA receptor insertion and a postsynaptic form of LTP are crucially important for fear learning. However, because a constitutive KO approach was used that inactivates GluR1 in a brain-wide fashion, it remains possible that the loss of GluR1 in other brain areas had contributed to the observed behavioral effects.
Plasticity at Cortical Inputs to the LA
As reviewed above, early studies provided evidence that besides the MGm/PIN, cortical areas such as the AuCx and the vAuCx/TeA project to the LA. Therefore, subsequent studies investigated long-term plasticity at the cortico-amygdala synaptic connection via electrical stimulation of the external capsule (Huang and Kandel 1998; Humeau and others 2003). As a potential caveat, it should be noted that external capsule stimulation is expected to activate inputs from various cortical sources rather than inputs from the AuCx alone. Nevertheless, we operationally refer to data obtained with external capsule–stimulation as the “cortico-amygdala synapse” (Fig. 2B). LTP can be induced in this pathway (Huang and Kandel 1998), and prior fear learning in rats leads to the partial occlusion of LTP, suggesting that plasticity at this synapse contributes to fear memory formation (Tsvetkov and others 2002).
A recent study tested the role of plasticity at the inputs to the LA from the AuCx and the thalamus in discriminative fear learning (Kim and Cho 2017). The study used Fos-CreERT2 mice (Guenthner and others 2013) to drive Cre expression by previous exposure to sounds, and it employed this to express channelrhodopsin selectively in those presynaptic auditory neurons that coded for the specific sound frequency (CS+ versus CS-). The study demonstrated that optogenetically evoked EPSCs at CS+ coding synapses to the LA arising from the AuCx and MGm/PIN had an increased AMPA/NMDA ratio following discriminative fear learning. Conversely, inputs coding for CS– were not potentiated. Potentiation of inputs coding for the CS+ was also observed when the expression of channelrhodopsin was restricted to the auditory cortical areas. Thus, the study showed that the synapses from the auditory cortical areas play a role in discriminative fear learning; furthermore, it demonstrates that synaptic plasticity is specific for those synapses that code for the relevant sound frequency.
The importance of synaptic plasticity at cortical and thalamic input synapses to the LA in fear learning was demonstrated with an elegant in vivo optogenetic stimulation approach to induce either LTP or long-term depression (LTD) in the two pathways together (Nabavi and others 2014). In rats that had already undergone fear conditioning, optogenetic in vivo stimulation of thalamic and cortical afferents with an LTD-inducing stimulus led to the disappearance of auditory-cued fear memory. Subsequent optogenetic stimulation of the same axons with an LTP-inducing stimulus then reinstated the fear responses upon CS presentation (Nabavi and others 2014). Interestingly, however, inducing LTP in naive rats was not able to create an artificial fear memory. This might indicate that for creating a fear memory, additional plasticity-inducing factors related to the natural transmission of “US information,” for example the activation of neuromodulatory systems, are necessary. Another study demonstrated that pairing of optogenetically activated inputs from the MGm and the vAuCx with a footshock, followed by optogenetic activation of the inputs alone 1 d later, led to the recall of an artificial “auditory”-cued fear memory. These experiments thus showed that optogenetic activation of the thalamic and cortical pathways to the LA can serve as an internally activated CS (Kwon and others 2014).
Fear memories can undergo extinction upon repeated exposure of fear-conditioned subjects with the CS alone (Milad and Quirk 2012; Orsini and Maren 2012). There seems to be a consensus that extinction is mediated by influences originating in the prefrontal cortex, which finally act by inhibiting output structures of the amygdala, both via inhibitory neurons in the intercalated cell masses of the amygdala (Likhtik and others 2008), and via activating local inhibition in the LA (Lin and others 2009; Rosenkranz and others 2003; for reviews see Milad and Quirk 2012; Orsini and Maren 2012). Interestingly, it has also been shown that fear extinction causes a depotentiation of EPSCs at the thalamic and cortical inputs to the LA after these synapses had been potentiated by previous fear learning (Hong and others 2009; Kim and others 2007). More recently, the same group found that the depotentiation of EPSCs at the thalamo-amygdala input continued over several extinction sessions, whereas the former inhibition-like mechanisms were more relevant when extinction training was brief (An and others 2017). Thus, a depotentiation of thalamic and cortical inputs by LTD-like processes might become more relevant for long-lasting extinction processes. These studies, together with the study by Nabavi and others (2014), thus demonstrated that the plasticity state at thalamic and cortical inputs to the LA are an important determinant of the level of fear memory expression (An and others 2017; Kim and others 2007).
The specific afferent synapses arising from the thalamic versus cortical area inputs to the LA have been visualized through a dual eGRASP approach (enhanced GFP [green fluorescent protein] reconstitution across synaptic partners; Choi and others 2018). This approach relies on the expression of split components of GFP in the pre- and postsynaptic neuron separately, and on the complementation of the split fluorescent proteins via contact in the synaptic cleft (Feinberg and others 2008). Choi and others (2021) used the expression of additional fluorescent proteins in a cFos- and doxycycline-dependent manner to define pre- and postsynaptic neuron populations that belong to a fear memory engram (see also Han and others 2007). They then quantified the volumes of the labeled postsynaptic spines in the LA as a proxy for LTP (Choi and others 2021). The study found that auditory-cued fear learning led to a significant ~50% increase in the volumes of spines between engram cells in the auditory cortical areas, and in the LA. Interestingly, extinction partially reversed the increased spine head volumes at the cortical-LA pathway, a finding that agrees with the depotentiation hypothesis of extinction (see above; An and others 2017). On the other hand, the thalamic inputs onto LA neurons did not result in a significant increase in spine volumes following fear learning. However, it was argued that technical limitations, such as a low number of presynaptic engram cells in the MGm/PIN, could have masked similar effects in the thalamic pathway, so these findings should not be used to conclude that fear learning goes along without plasticity in the thalamus to LA pathway (Choi and others 2021).
But How Is the “US Signal” Transmitted to LA Neurons?
The studies that we have reviewed so far present strong evidence that LTP occurs at synapses from the MGm/PIN to the LA, as well as at inputs from the auditory cortical areas, and that these processes are causally involved in the creation of an auditory-cued fear memory. These studies have led to the formulation of a widely accepted model of amygdala plasticity and fear learning. It postulates that activity at synapses that carry information about the CS, coincides with action potential firing in LA neurons driven by excitatory synapses carrying information about a footshock (US). Indeed, it was shown that a subpopulation of LA neurons is strongly activated by footshocks (Gore and others 2015; Grewe and others 2017). Then, a process of associative plasticity would strengthen the “CS-coding” synapses, which would allow for a stronger response of LA neurons to the learned CS after fear learning has occurred (Sigurdsson and others 2007; Fig. 4A). Nevertheless, newer findings question some assumptions of this model. First, the thalamic and cortical inputs discussed earlier might not selectively code for the CS but carry US information as well (Barsy and others 2020; Dalmay and others 2019; Taylor and others 2021). Also, it remains unclear whether there are separate glutamatergic afferents that carry strong US information to the LA as postulated in many models of associative plasticity for fear learning (Sigurdsson and others 2007). Alternatively, US information carried by inputs from the MGm/PIN and the vAuCx might be sufficient to drive US-evoked plasticity in the LA (Fig. 4B).
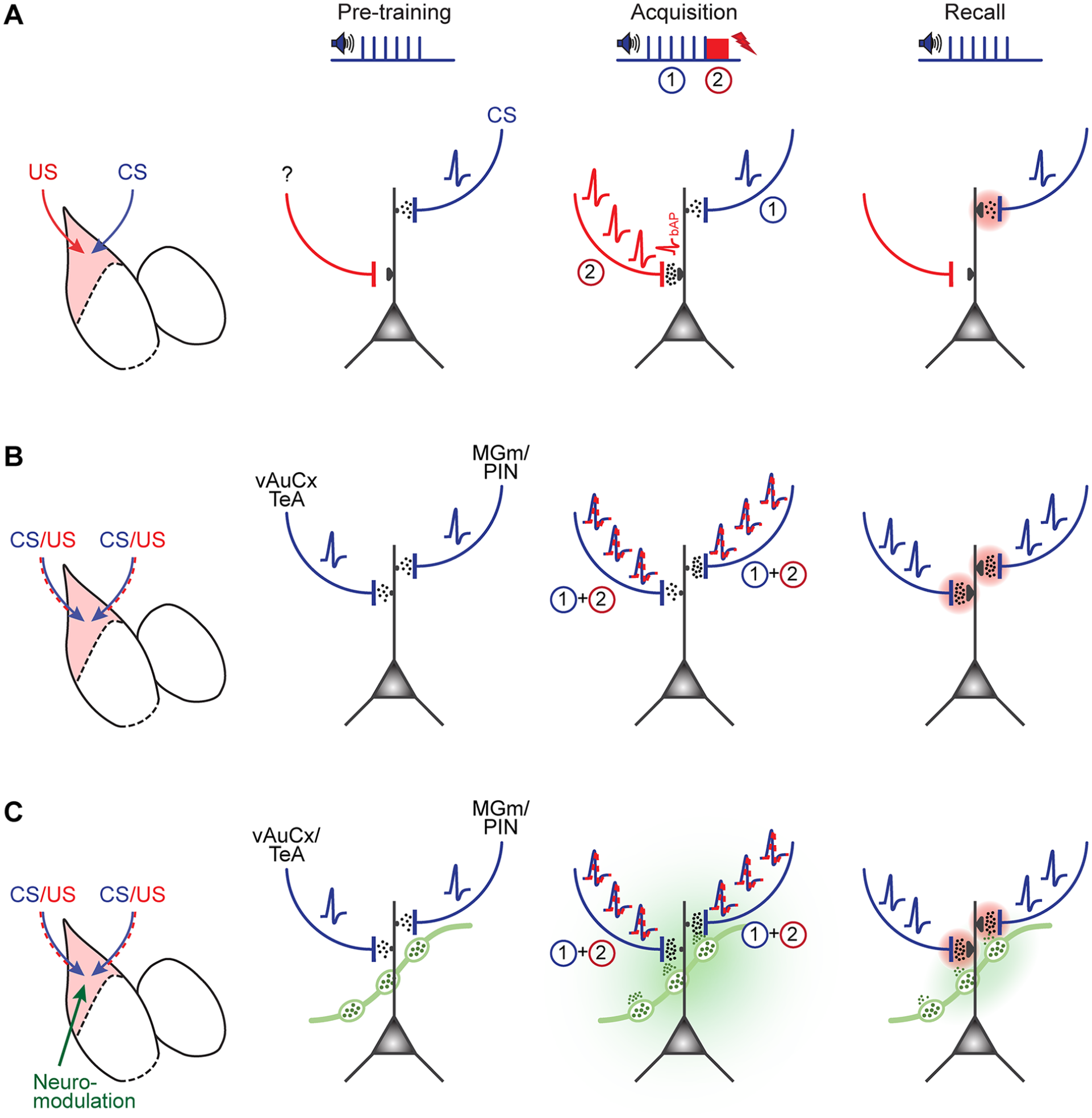
Transmission of conditioned stimulus (CS) and unconditioned stimulus (US) information in different models of fear learning–related plasticity in the lateral amygdala (LA). (A) Illustration of the classical model of associative plasticity for auditory-cued fear learning, as proposed by Sigurdsson and others (2007), based on many of the findings reviewed here. Note that CS information is assumed to be carried by afferents from the medial geniculate nucleus/posterior intralaminar nucleus (MGm/PIN) and the auditory cortical areas to the LA, whereas a so-far unidentified glutamatergic afferent is assumed to carry footshock information to the LA. As a result of coincident activity during fear memory acquisition, long-term potentiation occurs at the MGm/PIN input (pink shading in right panel). (B) An updated model takes into account that presynaptic afferents from the MGm/PIN and the ventral auditory cortex/ temporal association cortex (vAuCx/TeA) carry CS information on the pretraining and recall days, as well as mixed CS-US information on the fear learning training day. Note that during the training day, CS and US information occurs at slightly different times, as indicated by “1” and “2” (see panel A, Acquisition column). (C) As in panel B but now considering the release of neuromodulators such as noradrenaline and dopamine (green) during the memory acquisition phase. Because noradrenaline and dopamine neurons in the locus coeruleus and ventral tegmental area (not shown) acquire an action potential firing response to the CS after fear learning (Tang and others 2020; Uematsu and others 2017), it is possible that the neuromodulators are also released, albeit in lower amounts, during fear memory recall (right).
A series of studies has been performed to address the source of US information to the amygdala. Earlier lesion studies investigated whether the MGm/PIN might provide footshock information relevant for fear learning to the LA but finally concluded that this is not the case (Lanuza and others 2004). The parabrachial nucleus is a waystation in a spinomesencephalic pathway of nociceptive information toward higher brain areas (Gauriau and Bernard 2002). A series of studies found that footshock information is coded by CGRP-expressing neurons (calcitonin gene-related peptide) in the parabrachial nucleus, which make excitatory synapses onto CGRP receptor–positive neurons in the lateral and central parts of the CeA (Han and others 2015; Sato and others 2015). Optogenetic activation of this pathway causes freezing and has negative reinforcing qualities that can drive auditory-cued fear learning in mice (Han and others 2015; Sato and others 2015). Nevertheless, the parabrachial nucleus does not project to the LA. Taken together, the question is still unsettled whether there is a separate, presumably glutamatergic input that drives learning in the LA by depolarizing LA neurons during the footshock or an altogether different mechanism to induce plasticity (Herry and Johansen 2014).
We have so far conceptualized the US pathway as a bottom-up pathway, in which nociceptive information flows from lower to higher brain areas in the form of a teaching signal embodied by strong depolarization to induce plasticity at CS-coding synapses. Interestingly, learning theories posit that the prior expectation of a painful signal leads to a decreased neuronal representation of a US signal and to a decreased potential to induce learning (Fanselow 1998; Rescorla and Wagner 1972). Accordingly, it has been observed that repeated footshock stimuli result in decreasing responses in neurons of the periaqueductal gray (PAG) and the LA (Johansen, Tarpley, and others 2010). Although the PAG does not directly project to the LA, muscimol-induced inactivation of the PAG led to a reduction of US responses of LA neurons. A follow-up study indicated that a descending pathway from the CeA via the PAG and the brainstem provides feedback information for inhibiting expected aversive signals (Ozawa and others 2017). Along the same lines, an independent study in mice found that plasticity in the LA, measured as an increased AMPA/NMDA ratio at the optogenetically activated thalamic inputs to the LA, depends on activity of PKC-delta–positive neurons in the CeA (Yu and others 2017). In this case, a feedback circuit from the CeA via midbrain dopamine neurons and back to the LA was postulated; nevertheless, the functional role of this circuit has remained unaddressed and should be investigated in future work. Taken together, these studies showed that descending pathways from the CeA via the midbrain and other brain areas can set the strength of a “teaching signal” for plasticity in the LA.
Evidence for Mixed CS-US Transmission at Afferents to the LA
Two recent studies have re-addressed the role of the MGm/PIN in fear learning and reported that this brain area and its outputs to the LA already contain some information about aversive footshock. By using in vivo Ca2+ imaging with a miniature microscope, one study demonstrated that MGm/PIN neurons respond to tones and footshock and that during auditory-cued fear learning, a strengthening of tone responses occurs in a subpopulation of these neurons; however, the population response was unchanged (Taylor and others 2021). Second, Barsy and others (2020) used a Calretinin-Cre mouse, which targets a population of neurons within the MGm/PIN complex that projects to the LA (note that this study refers to the MGm/PIN area as “lateral thalamus”). Optrode recordings indicated that neurons in the MGm/PIN respond to tones as well as to footshocks and that these neurons increase their response to the CS during fear learning. Furthermore, optogenetic silencing of the MGm/PIN axons in the LA throughout the CS-US pairings led to a ~60% reduction of learned freezing during fear memory recall (Barsy and others 2020). These studies showed that during fear learning, neurons in the MGm/PIN acquire responsiveness to an aversively motivated tone (the CS), thus shining new light on older experimental evidence (Han and others 2008; Weinberger 2011).
These studies also suggest that the simple assumption in the “classical” model—that pure CS and US information becomes integrated in the LA—does not hold, at least not for the MGm/PIN-to-LA synapse. Rather, neurons upstream of the LA respond to tones and footshocks (Barsy and others 2020; Taylor and others 2021), implying that their output synapses carry both types of sensory information. It therefore appears that the synapses are strengthened by an LTP mechanism (Clem and Huganir 2010; Rumpel and others 2005), but information transfer is likely additionally increased because of increased presynaptic spike rates in response to a learned CS. This would mean that plasticity can be (partially) inherited from upstream brain areas, as suggested earlier (Cahill and others 1999). A similar argument might hold for the inputs from the vAuCx/TeA (Dalmay and others 2019). Taken together, models for associative plasticity should consider a mixing of CS and US information already in brain areas upstream of the LA (Fig. 4B).
The interaction between thalamic and cortical inputs onto LA neurons has been investigated in vitro, and postsynaptic spines contacted by each type of afferent have been identified by Ca2+ imaging. Both types of synapses are located in an intermingled fashion on dendrites of LA principal neurons; dendritic spines that receive thalamic input are larger than spines receiving cortical inputs (Humeau and others 2005). Interestingly, brief (~1 s) synchronous activation of thalamic and cortical presynaptic fibers leads to LTP in the cortical inputs, caused by glutamate spillover from thalamic axons and a subsequent increased glutamate release from the cortical inputs (Humeau and others 2003). This form of heterosynaptic associative plasticity might receive new relevance, since, as discussed earlier, MGm/PIN inputs and vAuCx/TeA inputs are activated by footshocks as well (Dalmay and others 2019; Barsy and others 2020; Taylor and others 2021). Furthermore, it is possible that inputs from additional cortical areas carry a similarly mixed CS-US signal to the LA. In this respect, the posterior insular cortex appears relevant, a cortical area that was shown anatomically to make connections to the LA (Shi and Cassell 1998; Gehrlach and others 2020) and is known to be activated by footshocks and other somatosensory signals, as well as by auditory stimuli (Rodgers and others 2008; Sawatari and others 2011).
Part of the US Signal Is Provided by Neuromodulatory Systems
A series of recent studies has provided evidence that neuromodulators play additional roles in signaling US information to brain areas such as the LA and other higher integration centers (see Likhtik and Johansen 2019 for a review). A study addressing the mechanisms inducing plasticity during fear learning found that optogenetically triggered depolarization of LA neurons, when paired with auditory (CS) presentation, is not fully efficient to induce associative fear learning (Johansen, Hamanaka, and others 2010). A follow-up study demonstrated that depolarization and activation of beta-adrenergic receptors in the LA together allow for an efficient induction of associative learning (Johansen and others 2014; Fig. 4C).
Studies reported that the activation of noradrenergic neurons in the locus coeruleus is a necessary factor for efficient fear learning (Uematsu and others 2017). Optogenetic inhibition of tyrosine hydroxylase–expressing locus coeruleus neurons during the footshock in a tyrosine hydroxylase–Cre rat model caused a ~50% reduction of learned fear (Uematsu and others 2017). In another study, the release of acetylcholine from the basal forebrain in the LA/BA was necessary for efficient fear learning (Jiang and others 2016). Finally, Tang and others (2020) identified a subpopulation of dopamine neurons by the use of dopamine transporter (DAT) Cre mice, which project to the BA. In vivo optrode recordings indicated that VTA neurons, among them DAT+ dopamine neurons, responded to footshocks and acquired a tone response during fear learning. Optogenetic silencing of VTA dopaminergic neurons during the footshock led to a moderate (~30%) reduction of fear memory; furthermore, silencing of VTA dopamine axons over the BA during the footshock reduced fear learning to a similar degree (Tang and others 2020). This study thus identifies a specific population of VTA dopamine neurons that projects to the BA, and that is involved in aversive learning. An interesting aspect is that both VTA dopamine neurons (Tang and others 2020) and noradrenergic locus coeruleus neurons (Uematsu and others 2017; see also Martins and Froemke 2015) acquire a response to the CS during fear learning. This observation suggests that some degree of dopamine and noradrenaline release might also occur in the LA and/or BA upon fear memory recall (Fig. 4C, right); the significance of this acquired neuromodulator release for processes of fear memory retrieval should be further investigated. Together, the aforementioned studies suggest that several classical neuromodulators are released in the LA and/or the BA during footshock presentation, where they likely gate synaptic plasticity leading to the formation of a fear memory. The cellular and circuit mechanisms of how the neuromodulators facilitate the induction of synaptic plasticity has not been investigated in detail. For exogenously applied dopamine, actions have been described on local inhibitory interneurons (Bissiere and others 2003) and on second messenger pathways in principal neurons involved in synaptic plasticity (Li and others 2011).
In Vivo Signatures of Associative Learning
As mentioned earlier, a classical model of associative plasticity postulates that during fear learning, the responses of LA principal neurons to the CS are increased (Sigurdsson and others 2007), and indeed early experiments found such increases of tone-evoked responses (Amano and others 2011; Quirk and others 1995; Rogan and others 1997). How fear learning might change the activity of populations of neurons in the LA and BA during and after fear learning has recently been reinvestigated (Grewe and others 2017; Zhang and Li 2018). Both studies used in vivo miniaturized microscope Ca2+- imaging (Ghosh and others 2011) to follow the activity of 100 to 200 neurons in a given mouse during repeated behavioral sessions. Grewe and others (2017) used a discriminative fear learning assay and reported that ~10% of the neurons responded to the CS+ under baseline conditions; this percentage increased to ~15% after fear learning. Interestingly, only about half of the neurons that increased their response to the CS+ also demonstrated a footshock (US) response on the fear learning training day (Grewe and others 2017). This surprising observation does not seem to agree with the postulate of coincident pre- and postsynaptic activity in classical models of associative learning. Moreover, the authors noted that among those neurons that responded to the CS, approximately one-third increased their CS response after fear learning (consistent with models of associative plasticity), whereas another third decreased their CS response (Grewe and others 2017). The authors then derived population vectors over the entire population of neurons and showed that fear learning shaped the population response to the CS+ to make it more similar to that to the US. On the other hand, the population response to the CS– was unchanged. Regarding the coexistence of neurons with increased and decreased CS responses, it is noteworthy that LA/BA neurons with different projection targets have been shown to display opposite plasticity after fear learning (LTP vs. LTD; Namburi and others 2015). Therefore, it might be questioned whether it is useful to integrate the entire population of LA/BA neurons into one “population response,” given that neuronal subpopulations can connect to different downstream targets and therefore might fulfil different roles in fear memory retrieval. The study by Grewe and others (2017) will be inspiring to theoretical studies of learning mechanisms. Furthermore, similar types of measurements could be made in future work imaging genetically or projection-defined neuronal populations in the LA/BA (see also Zhang and others 2021).
Serial Plasticity over Several Layers of Brain Organization
How does plasticity at excitatory synapses onto LA neurons finally create a memory that can elicit a defensive behavior, and how does an initially innocuous sensory percept (e.g., a tone) acquire an “emotional” value? To consider this question, we should look at the output connections of LA neurons, which are known to project to the BA and the CeA among other targets (Pitkänen and others 1995). Optogenetically assisted circuit mapping revealed a glutamatergic connection from the LA onto somatostatin-positive neurons of the lateral CeA, and ex vivo measurements revealed that fear learning drives a presynaptic form of LTP at this connection; moreover, optogenetic stimulation of somatostatin-positive neurons in the CeA causes freezing in naive mice (Li and others 2013). Although CeA circuits consist of complex inhibitory and disinhibitory loops (Ciocchi and others 2010; Haubensak and others 2010), these findings indicate that information about an auditory stimulus, arriving from thalamic and cortical sources, can gain access to the CeA by way of plastic changes at excitatory synapses originating in the LA. Furthermore, considering that MGm/PIN neurons acquire CS responsiveness and undergo learning-related changes (Barsy and others 2020; Taylor and others 2021); that LA neurons show plasticity at their input synapses from the MGm/PIN and auditory cortical areas (Kim and Cho 2017; Rumpel and others 2005), and finally, that the output synapses from LA principal neurons undergo LTP in the CeA (Hartley and others 2019; Li and others 2013), it is seen that LTP of synaptic transmission at long-range excitatory connections takes place over several brain layers (Fig. 5). This view, based on the studies reviewed here, significantly expands the classical model of associative plasticity, which postulates that CS and US information is integrated in a single layer of brain organization (Sigurdsson and others 2007; Fig. 4A).
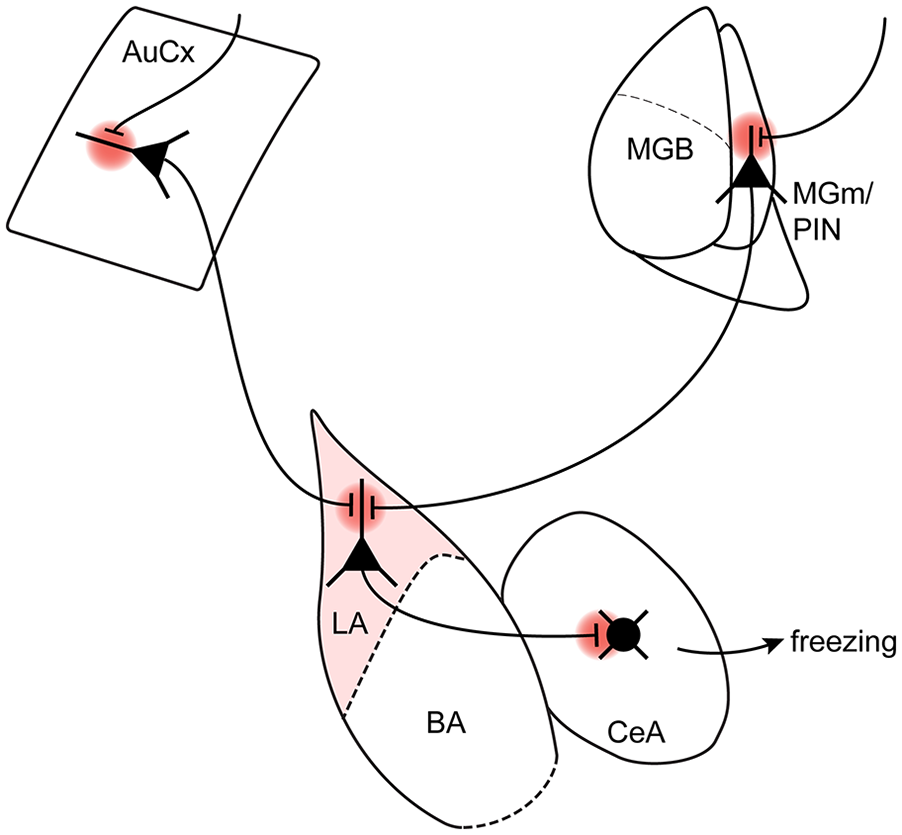
Plasticity happens in serial and parallel networks over subsequent layers of brain organization. See Figure 2 for abbreviations.
What might be the advantage for aversively motivated serial synaptic plasticity taking place over several layers of brain organization? Decades of research have shown that computation in sensory systems spans several brain layers (Felleman and Van Essen 1991), a finding that is exploited in machine learning by the use of convolutional neuronal networks (Yamins and DiCarlo 2016). It might be argued that a form of “all or none” plasticity within a single layer of brain organization, allowing a sensory representation to gain control over a strong defensive behavior such as freezing, would not enable a sufficiently flexible and fine-tuned control of a learned behavior. On the other hand, when synaptic plasticity takes place over several brain layers, its effects likely multiplicate, so a sensory percept of a CS can finally gain a sizable control over a behavioral decision. In addition, plasticity can now be regulated at several brain layers, which might allow for a more fine-grained control of the learned behavior (see discussion by Li and others 2013, who proposed a similar view). Optogenetic inactivation of individual brain areas aimed at suppressing localized CS-US integration has mostly reduced the amount of fear memory only partially; we give specific numbers reported in the original papers throughout. These findings indicate redundancy in the neuronal networks undergoing plasticity after fear learning, which is likely caused by serial arrangements of neurons undergoing aversively motivated plasticity. Additionally, parallel pathways are likely present, as exemplified by the two-tiered “auditory” inputs impinging from thalamic and cortical areas to the LA (Fig. 5). Thus, recent findings suggest a view in which synaptic plasticity in aversively motivated learning happens at several brain layers and that CS and US information is mixed earlier than previously thought (Barsy and others 2020; Dalmay and others 2019; Li and others 2013; Taylor and others 2021; Fig. 4B, C). Future theoretical work might embrace this view of plasticity.
Conclusions
Taken together, classical work showed that postsynaptic forms of plasticity and AMPA receptor cycling at thalamo- and cortico-amygdala connections are an important substrate for auditory-cued fear learning (Kim and Cho 2017; McKernan and Shinnick-Gallagher 1997; Nabavi and others 2014; Rumpel and others 2005). Nevertheless, more recent work has found that aversively motivated plasticity likely takes places in multiple brain areas both upstream and downstream of the LA. Furthermore, the development of the in vivo activity of amygdala neuronal populations during and after fear learning is more complex than predicted by a simple model of associative plasticity (Grewe and others 2017; Zhang and Li 2018). Finally, the synaptic afferents that might drive a subpopulation of LA neurons in response to US stimulation have not yet been identified, or they might be embodied in a mixed CS-US transmission to the LA; release of neuromodulators additionally plays a significant role as a teaching signal for plasticity in amygdala circuits (Johansen and others 2014). Many of these findings should be relevant for theoretical work on synaptic learning mechanisms (Gerstner and others 2018).
Considering these recent insights will help to derive new questions driving future research. We estimate that the paradigm of fear learning will continue to provide interesting new avenues into research linking synaptic plasticity mechanisms to neuronal network function and, eventually, to a more complete understanding of the mechanisms that underlie the formation and retrieval of fear memories.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Work on the mechanisms of fear learning in the authors’ laboratory has been funded by grants from the Swiss National Science Foundations (nº 31003A_176332/1, and 310030_204587/1, to R.S.).