
Abstract
Metabotropic glutamate receptors (mGluRs) are G-protein coupled receptors that are activated by glutamate in the central nervous system (CNS). Basically, mGluRs contribute to fine-tuning of synaptic efficacy and control the accuracy and sharpness of neurotransmission. Among eight subtypes, mGluR1 and mGluR5 belong to group 1 (Gp1) family, and are implicated in multiple CNS disorders, such as Alzheimer’s disease, autism, Parkinson’s disease, and so on. In the present review, we systematically discussed underlying mechanisms and prospective of Gp1 mGluRs in a group of neurological and psychiatric diseases, including Alzheimer’s disease, Parkinson’s disease, autism spectrum disorder, epilepsy, Huntington’s disease, intellectual disability, Down’s syndrome, Rett syndrome, attention-deficit hyperactivity disorder, addiction, anxiety, nociception, schizophrenia, and depression, in order to provide more insights into the therapeutic potential of Gp1 mGluRs.
Keywords
Introduction
Metabotropic glutamate receptors (mGluRs) have an extracellular ligand-binding domain, a heptahelical membrane structure, and a C-terminal tail. Different from ionotropic glutamate receptors, mGluRs belong to G-protein coupled receptors (GPCRs). Group 1 mGluRs (Gp1 mGluRs) consist of two members, mGluR1 and mGluR5. The locations of Gp1 mGluRs in the central nervous system (CNS) vary from isoform to isoform (Alvarez and others 2000). mGluR1 is mainly expressed in the hippocampus, cerebellum, and substantia nigra, meanwhile mGluR5 is expressed in the hippocampus, amygdala, olfactory bulb, striatum, nucleus accumbens, septum, and dorsal horn (Abe and others 1992; Hubert and others 2001; Jia and others 1999; Martin and other 1992; Romano and others 1995; Ryo and others 1993; Swanson and others 2005; Fig. 1). At synapses, mGluR1 and mGluR5 are mainly located at postsynaptic terminals (Fig. 2), although mGluR5 is also found at presynaptic site and mediates the depletion of phosphatidylinositol-4,5-bisphosphate (PIP2; He and others 2019).
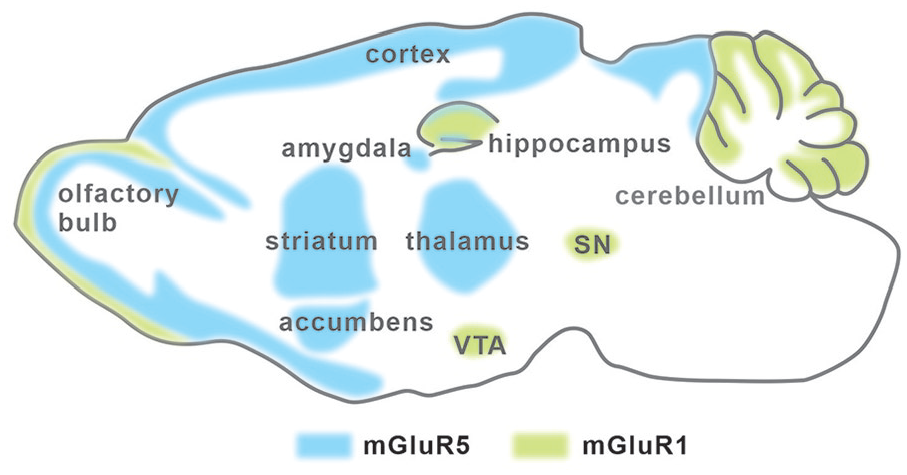
The expression of Gp1 mGluRs in CNS. mGluR1 expression is indicated by green shadow, and mGluR5 expression pattern is indicated by blue shadow. This figure is modified from Lüscher and Huber (2010). SN = substantia nigra.
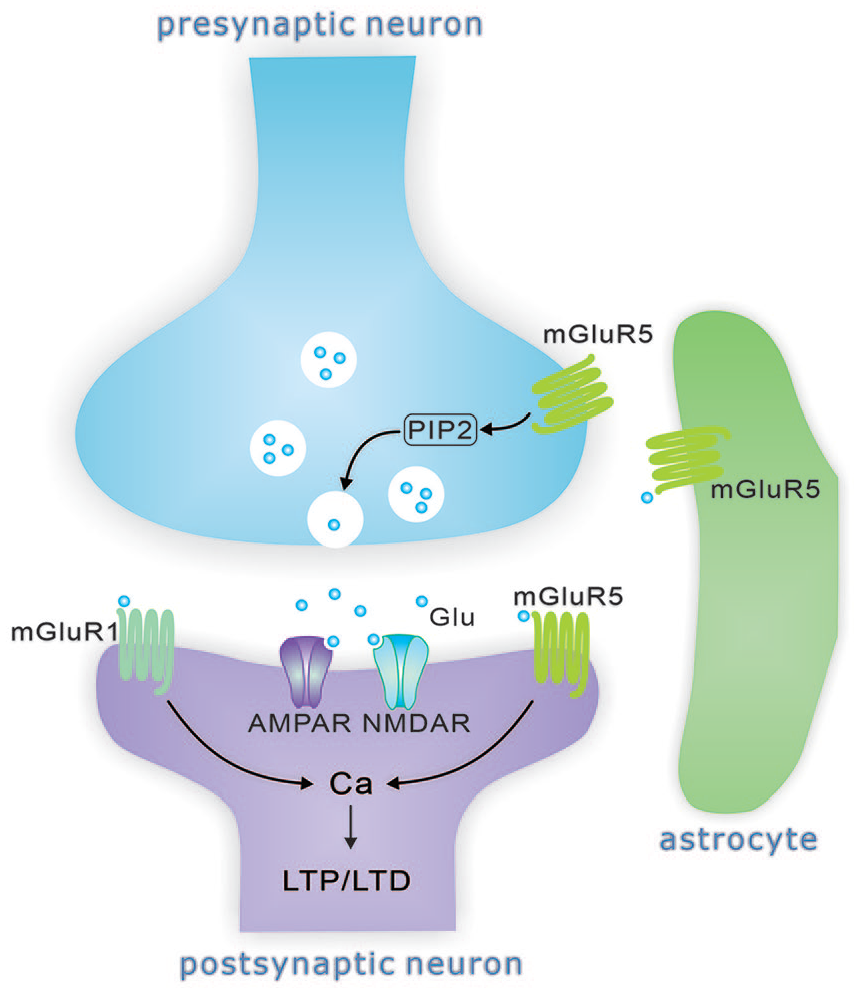
The distribution of Gp1 mGluRs at glutamatergic synapse. Gp1 mGluRs are mostly localized postsynaptically and promote a calcium influx upon their activation and contribute to LTP/LTD induction. mGluR5 also modulates synaptic release probability by depleting PIP2. The astrocytes also express mGluR5.
The domain of Gp1 mGluRs bears orthosteric binding sites for endogenous glutamate (Koehl and others 2019). In Purkinje cells, glutamate binding of mGluR1 exerts slow currents (Zhou and others 2017), which are produced by coupled transient receptor potential C (TRPC) channels (Kim and others 2003). Gp1 mGluRs are also subject to allosteric binding of certain modulators, such as positive allosteric modulators (PAMs) and negative allosteric modulators (NAMs; Feng and others 2015), which cause sensitization or desensitization of these receptors. PAMs and NAMs indirectly modulate glutamate transmission and are thereby used in clinical trials targeting related CNS disorders (Zoicas and Kornhuber 2019). Like other GPCRs, the activation of Gp1 mGluRs by glutamate or allosteric ligands causes conformational changes of extracellular domain and produces coupled G-protein signaling, which relays to multiple downstream molecules (Jojart and others 2020; Niswender and Conn 2010). Upon the binding of glutamate or modulators, Gp1 mGluRs activate phospholipase C-γ (PLC-γ), generate inositol triphosphate (IP3) and diacylglycerol (DAG), and trigger a local rise of intracellular calcium and the activation of protein kinase C (PKC; Maiese and others 2008). Through these mechanisms, they induce long-lasting changes of glutamatergic transmission, including long-term depression (LTD) and long-term potentiation (LTP), and neuronal excitability in cerebellar Purkinje cells, the hippocampus, neocortex, dorsal and ventral striatum, and spinal cord (Anwyl 1999; Ayala and others 2008; Bellone and others 2008; Gladding and others 2009; Jorntell and Hansel 2006; Kullmann and Lamsa 2008). Rich evidence demonstrates that Gp1 mGluRs function in synaptogenesis, neuronal development, learning and memory, and neurodegeneration (Conn and Pin 1997; Dale and others 2002; Nakanishi 1994; Pin and Duvoisin 1995; Pin and others 1994). The implications and mechanisms of mGluRs in several brain diseases have been reviewed within last decade (Amalric 2015; Kumar and others 2015; Lüscher and Huber 2010; Masilamoni and Smith 2018; Niswender and Conn 2010; Ribeiro and others 2010a; Ribeiro and others 2017; Srivastava and others 2020), focusing on individual diseases (mainly Alzheimer’s disease and Parkinson’s disease). In the present review, we will combine previous and more recent studies to discuss the underlying mechanisms and prospective of Gp1 mGluRs in not only Alzheimer’s disease and Parkinson’s disease but also autism spectrum disorder, epilepsy, Huntington’s disease, intellectual disability, Down’s syndrome, Rett syndrome, attention-deficit hyperactivity disorder, addiction, anxiety, nociception, schizophrenia, and depression.
Alzheimer’s Disease (AD)
Clinical studies reveal that levels of Gp1 mGluRs are reduced in the hippocampus and the cortex in postmortem brain of AD patients (Albasanz and others 2005), although one study shows no difference between AD and control groups at early stages (Ishibashi and others 2019). To clarify the controversy, longitudinal observations are required to determine the changes of Gp1 mGluRs associated with AD progression. Indeed, the reduction of mGluR1 occurs in cases with the progression of AD (Albasanz and others 2005). Moreover, reduced mGluR5 was found in 16-month-old Tg-ArcSwe mice, an AD mice model, when compared with control mice (Fang and others 2017), and in the hippocampus of mild AD patients when compared with age-matched controls (Mecca and others 2020).
The association of Gp1 mGluRs with AD progression has been investigated to a good extent. In AD, amyloid hypothesis holds amyloid-β (Aβ) peptide as the prime causative of AD pathology (Hardy and Selkoe 2002; Tanzi and Bertram 2005), as shown by disruptive effects of Aβ40 and Aβ42 on synaptic transmission and brain connectome (Crews and Masliah 2010; Holtzman and others 2012). Basically, it is speculated that Gp1 mGluRs mainly influence Aβ and amyloid precursor protein (APP), which creep synapse loss, cognitive decline, and silencing of brain circuits of AD (Overk and Masliah 2014; Palop and Mucke 2010). Still, the role of Gp1 mGluRs in AD is paradoxical: they may either prevent or promote the progression of AD depending on distinct conditions. The activation of Gp1 mGluRs accelerates non-amyloidogenic processing of APP and protects from Aβ-mediated neurotoxicity (Jolly-Tornetta and others 1998; Meziane and others 1998), suggesting that the activation of Gp1 mGluRs may be a potential strategy for reversing pathological changes in AD patients. In contrast to this finding, other studies demonstrated that Gp1 mGluRs and their upstream or downstream proteins together worsen AD progression. mGluR1 stimulation can increase APP secretion and leads to accumulation of Aβ (Nitsch and others 1997), which is mediated by the release of arachidonic acid via intracellular Ca2+ mobilization and ultimately triggers the formation of neurofibrillary tangles, a pathological hallmark of AD (Tsai and others 2005). More studies revealed the interaction between Aβ and mGluR signaling on neuronal function and structure. Soluble Aβ binds cellular prion protein (PrPC) during AD pathophysiology (Haas and others 2014), and Aβ-PrPC complex activates mGlu5 to disrupt neuronal function (Um and others 2013). Thus, mGlu5 is a co-receptor for Aβ binding to PrPC to disrupt normal neuronal signaling and function. This conclusion is confirmed by the finding that mGluR5 deletion improves AD pathogenesis and cognitive decline in APP/PS1 (Presenilin 1) mice (Hamilton and others 2014).
It appears that the change of Gp1 mGluRs-dependent synaptic plasticity is one culprit for cognitive impairment and dementia in AD patients. mGluR1-LTD is blocked in APP/PS1 mice and suppressing pERK (phosphorylated extracellular regulated protein kinase) reverses mGluR1-LTD failure (Yang and others 2016). mGluR1-LTD is facilitated by amyloid β (Aβ; Li and others 2009), which may explain the loss of structural and functional synapses or spines (Kamikubo and others 2006; Nagerl and others 2004; Shankar and others 2008) and the early learning and memory deficits in AD (Lüscher and Huber 2010). Indeed, Aβ causes mushroom spine loss by overacting mGluR5 in APP knock-in mouse model of AD (Zhang and others 2015). Moreover, mGluR-LTD is associated with shrinkage or loss of dendritic spines (Zhou and others 2004). Consequently, mGluR dysfunction results in compromised dendritic spines and synaptic memory (Ma and Klann 2012; Oddo and others 2003), and blocking this step may be a potential strategy for AD treatment. Yet the effect of Aβ on mGluR-dependent plasticity may be more complicated than expected. Exogenous Aβ leads to a removal of AMPARs that occludes mGluR-dependent LTD in cultured neurons (Hsieh and others 2006). Differently, Yang and others (2016) reported that mGluR-LTD is unaltered by Aβ application but blocked by APP/PS1 mutation. The experimental objects may explain the controversy. It is possible that an unknown mechanism compensates the short-term effect of Aβ on mGluR-LTD, which is yet impaired by chronic accumulation of Aβ with aging. In addition, a recent study provides evidence showing the role of mGluR5 at the early stage of AD: the release probability of hippocampal synapses is reduced in APP/PS1 mice due to mGluR5-mediated depletion of PIP2 (He and others 2019).
Accordingly, Gp1 mGluRs participate in AD by regulating synaptic function through a series of downstream and upstream signaling pathways. Therefore, some antagonists or modulators of Gp1 mGluRs have been taken in therapeutic treatment of AD. For example, 2-chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine (CTEP), a mGluR5-selective NAM (Lindemann and others 2011; Lindemann and others 2015), is proved to be effective for the treatment of AD, at least in APPs/PS1 mouse model of AD (Hamilton and others 2016). Unfortunately, very few medicines have perfect effect for AD due to side effects or low efficiency, although more and more clinical candidates related to Gp1 mGluRs are found. After all, Gp1 mGluRs are not the direct cause of AD while they play regulatory roles. This explains why Gp1 mGluRs-dependent medicines cannot provide the radical cure for AD. In fact, the immunotherapy that uses antibody to bind parenchymal Aβ becomes popular for the treatment of AD (Sevigny and others 2016). Still, unraveling cellular mechanisms of Gp1 mGluRs in AD helps discover a new medicine to prevent AD progression. For example, we reported that protein Numb and transferring receptor 1 (TFR1) are able to modulate the trafficking and surface expression of mGluR1 and mGluR5 (Wang and others 2019; Zhou and others 2015; Zhou and others 2017). Different from NAMs and PAMs, Numb and TFR1 do not interrupt Gp1 mGluRs in basal condition, instead they regulate the activity of mGluRs during neuronal plasticity. Hence, these molecules may be potential therapeutic targets for AD.
Parkinson’s Disease (PD)
PD symptoms are characterized by motor rigidity, bradykinesia, tremor, and postural instability, which largely arise through the progressive degeneration of dopamine neurons in the substantia nigra. These dopamine neurons are a key component of the basal ganglia, a highly organized network of brain nuclei implicated in motor, limbic, and cognitive functions. Gp1 mGluRs are expressed in basal ganglia nuclei, including dopaminergic neurons of substantia nigra, striatal projection neurons and interneurons, globus pallidus, and subthalamic nucleus (Conn and others 2005). Due to their expressions, the association between Gp1 mGluRs and PD was suggested two decades ago. It is acknowledged that the expression of both mGluR5 and mGluR1 is altered in the striatum and in the substantia nigra of PD models of non-human primate (Kaneda and others 2005; Morin and others 2013; Ouattara and others 2010; Samadi and others 2008; Sanchez-Pernaute and others 2008) and in parkinsonian patients with motor complications (Gregoire and others 2011). These findings were further strengthened by recent studies. Using positron emission tomography (PET), Kang and others (2019) showed that mGluR5 expression is increased in strategic dopaminergic brain regions of PD patients. PET imaging also shows that pathological progressions are accompanied by dynamic changes of mGluR1 in A53T transgenic rat model of PD (Yamasaki and others 2016). Moreover, mGluR5 knockdown was determined to decrease dyskinesia in an aphakia mouse model of PD (Garcia-Montes and others 2019). In addition, the binding potential of mGluR5 is decreased in 6-OHDA rat model of PD (Crabbe and others 2018).
Despite these findings, the precise roles of Gp1 mGluRs in PD yet remain unclear. One possibility is that Gp1 mGluRs influence basal ganglia synaptic transmission, as the activation of mGluR1/5 inhibits neuronal activity in the striatum (Blaabjerg and others 2003) and facilitates dopamine release from nigrostriatal terminals (Campusano and others 2002; Shimazoe and others 2002). Alternatively, Gp1 mGluRs cause Ca2+ oscillation and change the programming of transcriptional events (Bradley and Challiss 2011), which further enhances the activity of NR2B-containing NMDARs (Sarantis and others 2015). Moreover, the absence of mGluR-LTD results in a shift toward LTP within the striatum, and thereby leads to enhanced activity of indirect pathway and excessive inhibition of movements in PD (Kreitzer and Malenka 2007; Shen and others 2008). Thus, Gp1 mGluRs may participate in PD progression through changing the excitatory drive of basal ganglia nuclei.
Although the exact mechanism by which mGluR1/5 affect PD progression remains to be established, therapeutic implications of Gp1 mGluRs in PD have been substantially studied (Nickols and Conn 2014). In a 6-OHDA-induced rodent model of PD, subchronic intranigral administration of either LY367385, a mGluR1 antagonist, or MPEP, a mGluR5 antagonist, significantly slows down the degeneration of dopaminergic neurons, prevents 6-OHDA toxicity, and attenuates striatal dopamine depletion (Aguirre and others 2001; Battaglia and others 2002; Battaglia and others 2004; Vernon and others 2005; Vernon and others 2007). Follow-up studies corroborated these results, showing that MPEP attenuates parkinsonian motor deficits (Ambrosi and others 2010) and decreases dyskinesias in several animal models of PD (Dekundy and others 2006; Johnston and others 2010; Mela and others 2007; Rylander and others 2010). Systemic administration of 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl] pyridine (MTEP), another NAM of mGluR5, is also highly protective against MPTP-induced neurodegeneration in non-human primates (Masilamoni and others 2011). It was further indicated that MTEP treatment protects noradrenergic and serotonergic neurons of locus coeruleus and dorsal raphe against MPTP toxicity (Masilamoni and others 2011).
It appears that mGluR5 NAMs are more promising for the treatment of dyskinesia in PD patients compared to mGluR1. In support with this opinion, prevailing studies showed that mGluR5 antagonists have significant anti-dyskinetic effects in rodent and non-human primate models of PD (Dekundy and others 2006; Johnston and others 2010; Litim and others 2016) and some anti-parkinsonian effects in 6-OHDA-treated rats (Breysse and others 2003). More studies provide support toward the chronic use of mGluR5-related compounds as potential neuroprotective drug in PD: (1) The combined treatment with MPEP and L-DOPA significantly reduces dyskinesia intensity by ~70% in de novo MPTP monkeys (Morin and others 2013); (2) Fenobam, a mGluR5 NAM, reduces dyskinesias by ~50% in 6-OHDA rats and ~70% in MPTP monkeys (Ko and others 2014; Rylander and others 2010); (3) Dipraglurant, a mGluR5 NAM, reduces the severity of dyskinesias in MPTP macaque model (Bezard and others 2014) and in PD patients (Tison and others 2016); (4) mGluR5 antagonists (AFQ056-mavoglurant and ADX-48621-dipraglurant) show anti-dyskinetic functions and are well tolerated in human trials (Stocchi and others 2013). These preclinical studies provide evidence showing the effects of mGluR5 NAMs in PD therapy.
Autism Spectrum Disorder (ASD)
ASD patients are characterized by the deficits in social communication and social interaction, repetitive behaviors (American Psychiatric Association 2013), and motor behaviors (Gowen and Hamilton 2013). Although the pathogenic mechanism for ASD still remains elusive, increasing evidence suggests that Gp1 mGluRs are implicated in ASD according to the studies from ASD mouse models and patients (Auerbach and others 2011; Mercer and others 2016; Seese and others 2014; Wang and others 2016; Wenger and others 2016). Seese and others (2014) found that the density of pERK1/2 is abnormally low in the hippocampus of BTBRT+ ltpr3tf/J mice with autistic behaviors. Interestingly, MPEP can recover p-ERK1/2 content and rescue long-term memory deficit (Huang and others 2014), consistent with a previous study showing that mGluR5 increases neuronal ERK1/2 activity (Wang and others 2007). In Shank3 knockout mice, mGluR5-Homer scaffolding is impaired which leads to abnormal cortico-striatal circuit, defective learning, and ASD-like behaviors (Wang and others 2016). Deletion of 4E (eIF4E)-binding protein 2 (Eif4ebp2), which encodes 4E-BP2, leads to abnormal excitation-inhibition balance and ASD-like behaviors. The antagonists of mGluR1 (JNJ16259685) and mGluR5 (Fenobam) can recover social interaction and repetitive behaviors of Eif4ebp2-/- mice (Aguilar-Valles and others 2015). Gp1 mGluRs are also involved in ASD patients (Wenger and others 2016). Copy number variants occur more frequently in mGluR network (over 270 genes) of ASD children than controls (Hadley and others 2014). It is noteworthy that mGluR network genes are found in the 22q11.2 region and chromosome 21, which are involved in autistic behaviors in Down’s syndrome. These data suggest that a second hit derived from abnormal mGluR network may contribute to ASD phenotypes in patients with trisomy 21 or 22q11.2DS (Wenger and others 2016). Interestingly, the roles of Gp1 mGluRs in synaptic excitation may be sex dependent. Gabrb3 is one of the autism-linked genes expressed in the cerebellum (Hortnagl and others 2013). Gabrb3 deletion exerts different results in male and female mice: Neurons in mutant cerebellar nuclei of males rather than females display enlarged mGluR1/5 responses and accelerated spontaneous firing (Mercer and others 2016).
Some antagonists of Gp1 mGluRs are suggested to rescue ASD phenotypes in mouse models (Aguilar-Valles and others 2015; Belozertseva and others 2007; Molina-Hernandez and others 2008; Thomas and others 2012). JNJ16259685 restores impaired social behaviors only in Eif4ebp2 knock-out mice, but not in Shank2 knockout rats (Modi and others 2018). The question remains whether agonists or PAMs of mGluR1 might be efficacious in reversing social deficits in Shank2 knockout rats, given that Shank2 knockout mice show decreased NMDAR function. CFMTI, another selective mGluR1 antagonist, improves social interaction deficits induced by MK-801 in rats (Satow and others 2009), yet holding the potential of mGluR1 antagonists in reversing pathological social interaction. There seem differential actions of mGluR1 and mGluR5 on autistic behaviors. mGluR1 was characterized much less in the context of emotion and behavior but more cognitive dysfunctions in mouse models of ASD. In contrast, mGluR5 antagonists tend to act on cognitive deficits following high doses and their effect are less severe than that of mGluR1 antagonists.
Although pharmacological blockade of Gp1 mGluRs function emerges as potential therapeutic strategy for the treatment of ASD, overall there is still a huge gap for these antagonists to be ideal therapeutic drugs for ASD. Importantly, ASD is caused by a combination of genetic and environmental factors. Therefore, the treating with antagonists of Gp1 mGluRs possibly breaks ASD into individual symptom rather than as the whole. In this scenario, a combination of multiple treatments is needed to efficiently alter the progression of ASD.
Epilepsy
It has been acknowledged that stimulation of Gp1 mGluRs elicits ictal-like responses from normal hippocampus, which become persistent and show no fading even upon washout of the agonist (Zhao and others 2011; Zhao and others 2015). In fact, the long-lasting epileptiform discharges induced by Gp1 mGluRs on hippocampal network provides a model for the study of epileptogenesis. For example, increased expression of mGluR1/5 leads to epilepsy-associated focal cortical dysplasias and human temporal lobe epilepsy (Aronica and others 2003) and chronic seizures display in transgenic mice with the overexpression of mGluR1 (Pitsch and others 2007). It is noteworthy that the agonists of mGluR1 and mGluR5 are uniformly convulsant and their antagonists are conversely anticonvulsant (Moldrich and others 2003). The convulsant effects of Gp1 mGluRs agonists may be produced by inhibiting cationic nonselective channels or by potentiating NMDAR and/or AMPAR responses (Heidinger and others 2002). In addition, Gp1 mGluRs might be involved in epilepsy by either impairing GABAergic potential (Marino and others 2001) or enhancing presynaptic glutamate release (Cartmell and Schoepp 2000). The anticonvulsant effects of mGluR1/5 antagonists are found in several seizure rodent models. For example, mGluR1 antagonists, LY367385 and AIDA, robustly suppress generalized motor seizures (Chapman and others 1999; Moldrich and others 2003); potent antagonists of mGluR5, MPEP and SIB-1893, suppress clonic seizures at low doses (Chapman and others 2000). However, the effects of MPEP are rather complicated, as it suppresses neuronal firing triggered by DHPG at low doses (Gasparini and others 1999) but acts to modulate GABAergic transmission and norepinephrine receptors at high doses (Battaglia and others 2004; Mathiesen and others 2003).
A group of phenylglycine-like mGluR1/5 antagonists have demonstrated potent anticonvulsant activity in models of DHPG-induced limbic seizures. Intra-cerebroventricular administration is required for these phenylglycine derivatives to demonstrate anticonvulsant activity. A series of aminopyridine derivatives have been used as potent noncompetitive antagonists with the high selectivity for mGluR1. LY456236 is one of such compound and is shown to be anticonvulsant against sound-induced seizures, limbic seizures, focal seizure, and amygdala-kindled seizures (Shannon and others 2005). It is likely that there is no subtype difference in anticonvulsant actions, since the selective antagonists for mGluR1 and mGluR5 are equally potent and non-subtype-selective antagonists appear to be the most potent anticonvulsants (Merlin 2002; Stoop and others 2003). Therefore, a synergic interaction between mGluR1 and mGluR5 on neuronal bursts needs to be explored.
The underlying mechanisms for Gp1 mGluRs in epilepsy are still under investigation. PLC activation may be a critical component of epileptogenic process. Inhibition of PLC or PLCβ1 deletion prevents induction of ictal-like discharges (Chuang and others 2001). Furthermore, agents that interfere with Ca2+ release from intracellular stores prevent ictaform activity (McDonald and others 1993) suggesting that mGluR-induced epileptogenesis depends on intracellular Ca2+ mobilization. In addition, the inhibition of ERK1/2 phosphorylation prevents ictal-like discharges, suggesting that mGluR-induced epileptogenesis also depends on ERK 1/2 response (Zhao and others 2004).
Huntington’s Disease (HD)
HD is an autosomal-dominant neurodegenerative disorder caused by a poly-glutamine expansion in huntingtin (htt) protein. Primary symptoms of HD include chorea, loss of cognition, psychiatric disturbance, and death. Mutated htt is proposed to cause the progressive loss of neurons in the caudate-putamen and neocortex of HD patient’s brain. Ample evidence indicates that mGluR1/5 play essential roles in HD through interacting with htt protein or cooperating with NMDAR. On one hand, mGluR5 directly interacts with mutant htt protein (Anborgh and others 2005), which functions in a knockin mouse model of HD (Ribeiro and others 2010b). On the other hand, the activation of Gp1 mGluRs enhances NMDAR-induced membrane depolarization and intracellular Ca2+ accumulation in medium spiny neurons (MSNs; Calabresi and others 1999). Consequently, the stimulation of mGluR1/5 facilitates the sensitization of IP3 receptor and enhances Ca2+ release from intracellular stores in HD mouse models (Ribeiro and others 2010b; Tang and others 2003).
Likewise, the antagonists of Gp1 mGluRs, such as MPEP, have been used to treat HD mouse models. Basically, MPEP works well against HD: (1) MPEP increases survival time and improves rotarod performance of HD mice, though it does not decrease htt aggregate formation (Schiefer and others 2004); (2) the deletion of mGluR5 in a mouse model of HD (HdhQ111/Q111) improves rotarod performance and decreases the number of htt intranuclear inclusions (Ribeiro and others 2014); (3) decreased Ca2+ release and neuronal apoptosis are seen in cultured MSNs derived from HD mouse model when treated with MPEP (Tang and others 2003). However, other studies suggest that the neuroprotective effects of MPEP or MTEP are not mediated by mGluR5, because these antagonists elicit same effects to mGluR5 knockout neuronal cultures (Lea and others 2005). Moreover, Gp1 mGluRs may have a dual function to either promote neuro-protection or exacerbate neuronal death, depending on neuronal type and drug incubation paradigm (Bruno and others 2001). Overall, it is generally accepted that mGluR5 is important for HD-mediated alterations in locomotor behavior. This conclusion was corroborated by more experiments using mGluR1/5-related drugs. For example, CDPPB, one PAM of mGluR5, is capable of delaying HD-related symptoms in vitro and in vivo. Chronic treatment of CDPPB in BACHD mice (another HD mouse model) significantly activates cell signaling pathways important for neuronal survival, prevents neuronal loss in the striatum, and decreases htt aggregation (Doria and others 2015). In behaviors, CDPPB treatment is efficient to ameliorate motor incoordination and to rescue memory deficits exhibited by BACHD mice (Doria and others 2015). Importantly, no toxic effects or stereotypical behavior are observed upon CDPPB treatment (Doria and others 2015). These data indicate that CDPPB is a promising drug to treat HD.
It has been shown that several intracellular signaling molecules are involved in the action of mGluR1/5 in HD. First, the attenuation of mGluR1/5 signaling observed in HdhQ111/Q111 mice is PKC dependent and only present in young asymptomatic mice (Ribeiro and others 2010b). It is possible that PKC-mediated mGluR1/5 desensitization is protective, avoiding further increases in Ca2+ release that may result in increased cell death. Second, mGluR1/5 activates cell signaling pathways, including ERK1/2, PI3K/Akt, and mammalian target of rapamycin (mTOR), which contribute to neuroprotection. Indeed, mGluR5 activation leads to higher levels of ERK and Akt phosphorylation in cultured HdhQ111/Q111 striatal neurons (Ribeiro and others 2010b). The phosphorylation of Akt may function to reduce htt aggregate formation and neuronal cell death (Warby and others 2009). Third, CDPPB increases mRNA level of BDNF in the cortex while it decreases neuronal loss and htt aggregation (Doria and others 2015). Increasing BDNF level is probably an important neuroprotective mechanism activated by CDPPB, as BDNF promotes differentiation, plasticity, and survival of neurons in the cortex and striatum (Poo 2001). Indeed, behavioral performance is improved and disease progression slows down when BDNF is over-expressed in transgenic HD mice (Xie and others 2010).
Other Disorders
Gp1 mGlus are also involved in other disorders, including intellectual disability (ID), Down’s syndrome (DS), Rett syndrome (RTT), attention-deficit hyperactivity disorder (ADHD), addiction, anxiety, nociception, schizophrenia, and Renpenning syndrome.
Fragile X syndrome (FXS) is one of ID most studied and is caused by the expansion of CGG repeats in Fmr1 gene. Synaptic plasticity in the hippocampus has been mostly studied in Fmr1 knockout mice, and the results showed that NMDAR-dependent LTP and LTD, the prominent forms of plasticity, are unaltered in Fmr1 knockout mice (Huber and others 2002). Hence, research attention was moved to mGluRs-dependent synaptic plasticity and a definition of “mGluR theory” was derived from compelling in vitro and in vivo evidence in FXS (Bear and others 2004). mGluR5-dependent plasticity is exaggerated in Fmr1 knockout mice compared to wild type mice (Huber and others 2002), consistent with a fact that both Fmr1 deletion and mGluR5 stimulation result in increased protein synthesis. This finding led to a hypothesis that regulating mGluR5 activity may be beneficial for the impairment caused by Fmr1 deletion. In fact, crossing of Fmr1 knockout mice with mice heterozygous for mGluR5 induces the recovery of protein synthesis and is able to rescue aberrant dendritic spines (Dolen and others 2007). Therefore, many clinical trials have been aimed at the efficacy of mGluR5 antagonists or NAMs in FXS patients since then. Unexpectedly, most of trials failed to discover any benefit in the defective behaviors of subjects (Berry-Kravis and others 2018). Nevertheless, other efforts are still continued to verify whether these treatments could provide better results in individuals of different ages or within the duration of treatments. Besides FXS, Renpenning syndrome is a type of X-linked ID that shows microcephaly, short stature, small testes, and specific facial dysmorphism (Kalscheuer and others 2003; Rejeb and others 2011). The mutations of polyglutamine-binding protein 1 (PQBP1) gene in human are demonstrated to be associated with Renpenning syndrome (Ito and others 2009). A recent study demonstrated that the deletion of Pqbp1 interrupts eEF2K/eEF2 pathway and impairs mGluR-LTD and related behaviors (Shen and others 2021). This work not only reveals the underlying mechanism of Renpenning syndrome but also identifies PQBP1 as a new player in mGluR signaling and synaptic plasticity. Interestingly, both PQBP1 and FMRP regulate the activity of eEF2 and subsequent de novo Arc/Arg3.1 translation (Park and others 2008; Shen and other, 2021), suggesting that they may coordinately control Arc/Arg3.1-depenent mGluR-LTD and the progress of ID.
There are few studies on the role of mGluRs in DS. It is shown that mGluR5 expression is increased in the cerebral cortex of DS patients (Oka and Takashima 1999). Specifically, the expression of mGluR5 in hippocampal astrocytes increased at the middle phase of gestation and it maintained postnatally (Iyer and others 2014). However, there appears no correlation between mGluR5 increase and the number of astrocytes in DS patients: a study using same cohort of patients failed to detect difference in the expression of GFAP (Kanaumi and others 2013).
The investigation on the involvement of mGluRs in RTT is also few. Mecp2 knockout mice display an alteration in protein synthesis after the induction of mGluR5-LTD (Tao and others 2016). Further analysis on the genome-wide profile of ribosome-bound mRNAs demonstrates different expression in Mecp2 knockout mice: the majority of mRNAs are upregulated in knockout mice, suggesting that MECP2 acts as a repressor of gene expression. Most affected genes upon Mecp2 loss are involved in cytoskeleton organization and cell morphogenesis, which play key roles in the transport of synaptic vesicles, spine morphology, and dynamics (Tao and others 2016). NAMS for mGluR5 were used to chronically treat Mecp2 knockout mice and this treatment significantly reduced upregulated ribosome-associated mRNAs and improved phenotypes of Mecp2 knockout mouse (Tao and others 2016).
The role of mGluRs in ADHD is even just at the beginning. A whole-genome analysis demonstrated mutations in genes encoding for mGluRs: Grm5, Grm7, and Grm8 deletions and Grm1 duplication were found in several patients (Elia and others 2011), suggesting that up to 10% of ADHD cases may present variances in mGluR network.
mGluR1 and mGluR5 appear to be implicated in addiction via synaptic plasticity. mGlu-LTD may reverse synaptic potentiation of cocaine in ventral tegmental area (VTA), which is required for subsequent synaptic adaptations in the nucleus accumbens (NAc; Bellone and Lüscher 2006; Mameli and others 2009). mGluR1 needs to bind to Homer isoforms to induce LTD in the VTA. When mGluR1-Homer interaction is disrupted, the plasticity in response to single injection of cocaine becomes persistent and drives synaptic adaptations in the NAc (Mameli and others 2009).
mGluR5-knockout animals exhibited reduced level of anxiety (Brodkin and others 2002), and stress-induced hyperthermia was attenuated by MTEP in wild-type mice but not in mGluR5 knockout animals, highlighting the involvement of mGluR5 in anxiety (Brodkin and others 2002). In addition, mGluR5 expression was increased in CA1 but decreased in CA3 region of the hippocampus in depression, indicating that mGluR5 is possibly engaged in the mechanism of depression (Wieronska and others 2001).
mGluR5 may be involved in nociception following tissue injury (Walker and others 2001) and in brain ischemia (Bao and others 2001). In the middle cerebral artery occlusion (MCAO) experiment, both mGluR5 agonist (CHPG) and antagonist (MPEP) have neuroprotective effects with different mechanisms: CHPG may limit neuronal apoptosis after ischemia, while MPEP reduces neuronal damage and improves neurological recovery induced by ischemia.
Gp1 mGluRs are also implicated in schizophrenia. In the perinatal phencyclidine (PCP) model of schizophrenia, mGluR5 level is significantly increased in the prefrontal cortex and the hippocampus, whereas hippocampal mGluR1α is reduced (Lum and others 2016). Hence, the disrupted expression of mGluR1 and mGluR5 may underlie neurodevelopmental alterations after PCP treatment. In addition, the reduction of dysbindin-1, which occurs in the brains of schizophrenia patients, leads to impaired expression of Gp1 mGluRs, and the phosphorylation of ERK1/2 is also strikingly reduced during these processes (Bhardwaj and others 2015). Thus, dysbindin-1 may be another modulator of Gp1 mGluRs and the potential target for the treatment of schizophrenia.
Finally, recent work reveals the role of mGluR5 in depression, another psychiatric disease of great concern. First, PET scan indicated the decreased level of mGluR5 in several brain regions of patients with MDD (Deschwanden and others 2011). Second, antidepressant effects MPEP, MTEP, GRN-529, basimglurant, and DSR-98776 were demonstrated in animal models of depression (Chaki and Fukumoto 2018). Full NAMs of mGluR5, such as MTEP or MTEP, potentiated PCP-induced hyperlocomotion in rodents (Gould and others 2016), presumably due to the inhibition of NMDAR. Interestingly, partial NAMs of mGluR5 also exerted antidepressant effect without affecting PCP-induced hyperlocomotion (Gould and others 2016). Thus, partial NAMs may be the better clinical target for safe. Although no statistical effect was observed in the efficacy of basimglurant examined in patients with MDD, a trend toward a beneficial effect was found (Quiroz and others 2016). Therefore, further clinical studies are needed to evaluate mGluR5 NAMs as antidepressant drugs. The antidepressant effects of mGluR5 NAMs were not antagonized by TrkB inhibitor (Iijima and others 2012) or mTOR inhibitor (Iijima and others 2012; Palucha-Poniewiera and others 2014b), and mGluR5 NAM did not affect mTOR cascade or synaptic protein (Palucha-Poniewiera and others 2014b). Therefore, the neural mechanism for the antidepressant effect of mGluR5 NAMs is distinct from that for ketamine. Instead, the antidepressant actions of mGluR5 NAMs were blocked by depletion of 5-HT (Fukumoto and Chaki 2015; Palucha-Poniewiera and others 2014a). Thus, serotonergic transmission may be involved in the actions of mGluR5 NAMs.
Conclusions
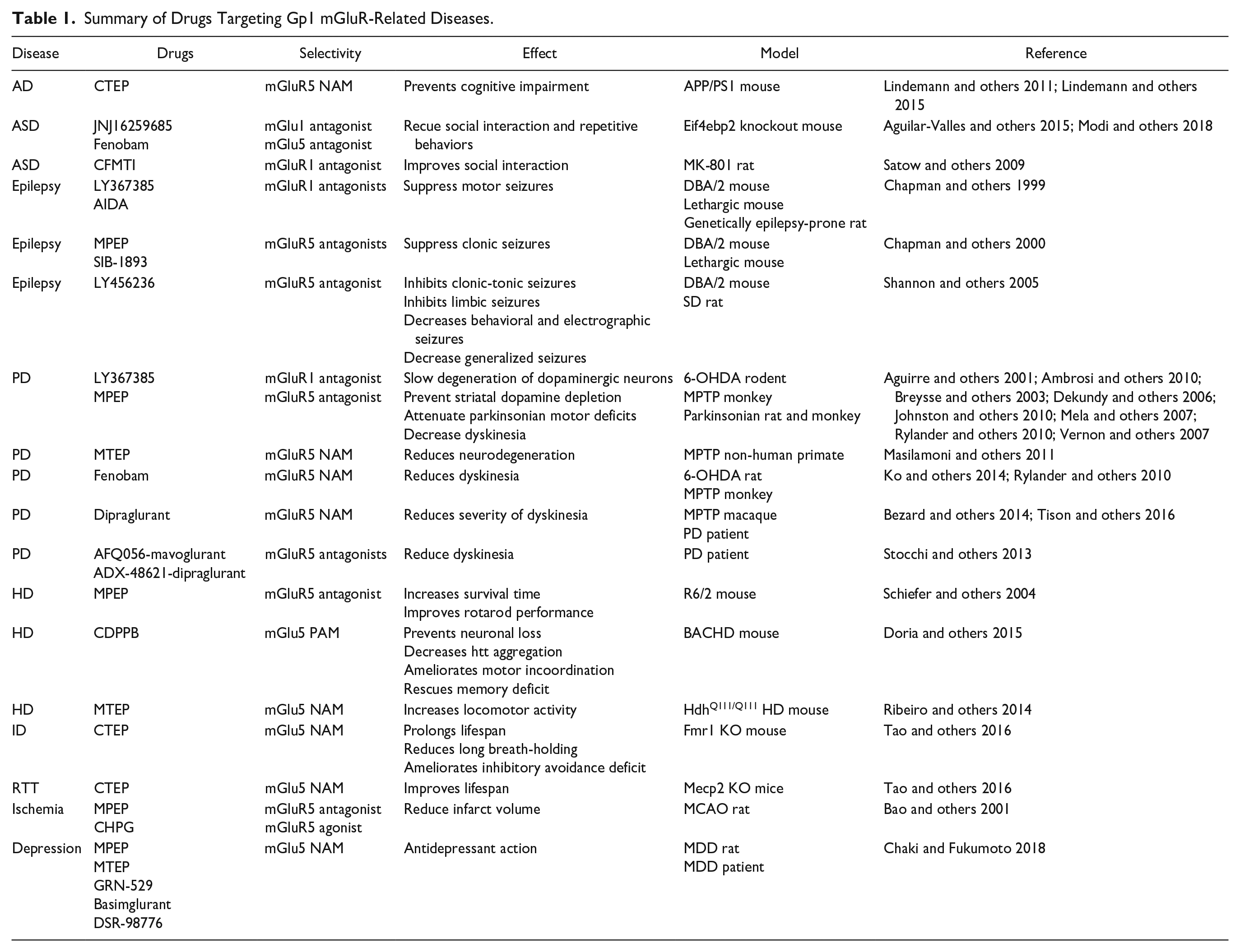
In this review, we discussed the roles of Gp1 mGluRs in the major neurodevelopmental, neurodegeneration, and psychiatric disorders. Some mechanisms are shared among different pathologies. However, determining whether the defects are similar direction among different disorders is more complicated due to the complexity of pathologies and animal models studied. Still, the development of specific pharmacological tools and transgenic animals has greatly advanced our understanding to the functions of Gp1 mGluRs in CNS disorders. Accordingly, the antagonists or NAMs of Gp1 mGluRs, such as MPEP or CTEP, have been suggested as treatments for different pathologies with promising effects. For convenience, we here listed kinds of agonists, antagonists, PAMs, and NAMs of Gp1 mGluRs that have been used in animal models and patients in Table 1. However, clinical trials still have controversial results, which may be biased by small number of patients. In addition, prevailing drugs mainly aim at the direct inhibition or activation of mGluRs, which may bring about many side effects. For example, Berry-Kravis and others (2018) summarized that PAMs and NAMs of mGluRs have been tested in comprehensive drug development programs undertaken for a wide range of neurodevelopmental disorder. However, none of the trials are able to unambiguously demonstrate efficacy in the drug development for FXS and other neurodevelopmental disorders (Berry-Kravis and others 2018). Still, we have very few knowledge to the role of Gp1 mGluRs on some diseases, such as RTT, ADHD, anxiety, schizophrenia, and motor incoordination. Further efforts are needed to better understand how the modulation of Gp1 mGluRs can be used in patients to ameliorate their symptoms, and more clinical trials on these diseases may bring us new hope.
Summary of Drugs Targeting Gp1 mGluR-Related Diseases.
Greater efforts have been made to achieve a general view on mGluR network, mainly cooperative proteins and receptors (Fig. 3). These identified signaling molecules provide new insights on the prospective of mGluRs in neurological disorders, although their implications in the pathologies are not sufficient enough, mainly due to technical and methodological limitations. Indeed, auxiliary proteins that modulate mGluRs activity in certain cellular processes may be better targets for related diseases. For example, transmembrane AMPA receptor regulatory protein (TARP), which regulates the trafficking of AMPARs, was considered to be a potential target in rescuing cognitive defects (Ishii and others 2020). Different from NAMs and PAMs, these molecules do not always boost or block Gp1 mGluRs in basal condition. Accordingly, several steps forward have been made with a long way to go before better understanding the complete picture.
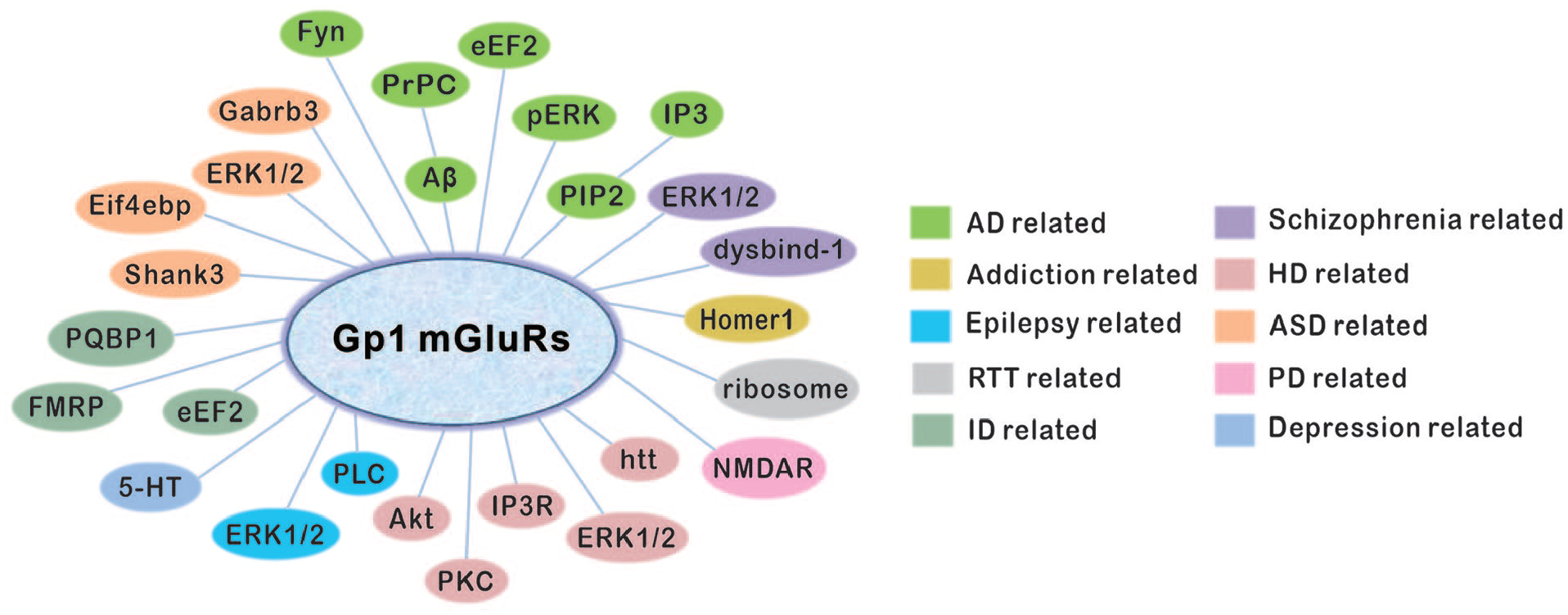
The upstream and downstream proteins of Gp1 mGluRs in a group of neurological and psychiatric diseases.
Footnotes
Acknowledgements
We thank Dr. Iain Bruce for polishing this manuscript and the members of Shen lab for their invaluable feedback on this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (81971874, 81625006, 31970923, and 31820103005).