
Abstract
The 22q11 deletion syndrome (22q11DS) is the most common microdeletion syndrome in humans and presents with a complex and variable psychiatric phenotype. Patients show cognitive impairment and have a higher probability of psychiatric disorders. As much as 30% of patients with 22q11DS suffer from schizophrenia, the strongest association between any mutation and the disease. Schizophrenia is a complex psychiatric disease that affects multiple brain regions, giving rise to a constellation of seemingly unrelated symptoms including hallucinations, social withdrawal, and memory deficits. Synaptic or neuronal malfunctions within certain physiological circuits appear to be at the core of these symptoms. Understanding disease at the synaptic level requires genetic model systems where intact neural circuits can be interrogated for functional deficits. Because of the overlap between 22q11DS and schizophrenia, models of 22q11DS may be key genetic tools for investigating both diseases. Here we discuss the advantages of using a synaptic function approach to studying mouse models of 22q11DS, review recent findings, and discuss them in the broader context of 22q11DS and schizophrenia.
22q11 Deletion Syndrome and Schizophrenia
Schizophrenia occurs in 1% of the world’s population and is characterized by positive symptoms (e.g., hallucinations, delusions), negative symptoms (e.g., loss of motivation, dampened emotions), and cognitive symptoms (e.g., deficits in working and long-term memory). Antipsychotic agents, most of which inhibit D2 dopamine receptors, can control positive symptoms with variable efficacy. However, they have many undesirable side effects and little effect on negative or cognitive symptoms (Keefe and Harvey 2012; Lieberman and others 2005). Since the introduction of antipsychotics in the 1950s, only a few new and valid pharmacologic targets for treating schizophrenia have been proposed.
The etiology and pathogenesis of schizophrenia remains unclear. Unlike other neural diseases such as Alzheimer’s or Parkinson’s disease, schizophrenia presents no discernible pathologic hallmarks to narrow studies to specific brain regions. As a result, almost every brain region and neurotransmitter system has been implicated in schizophrenia pathophysiology. Similarly, extensive genomic studies of schizophrenia patients and animal models have implicated various genes and cellular processes in the disease. However, links between implicated genes and functional deficits of the disease have yet to be made. Valid animal models of schizophrenia are needed to ascertain such links. However, because specific gene mutations predict schizophrenia at a very low rate, establishing such genetic models of the disease has been problematic.
The 22q11 deletion syndrome (22q11DS) is the most common microdeletion syndrome, occurring in approximately 1 in 4000 births (Oskarsdottir and others 2004). The deletion affects a 1.5- to 3-MB region of DNA carrying approximately 25 to 40 genes (Carlson and others 1997; Scambler and others 1991). Patients with 22q11DS face a range of physical and psychiatric outcomes, including predisposition to psychiatric disease (Fig. 1a) (Bearden and others 2001; Eliez and others 2000; Philip and Bassett 2011; Swillen and others 2000). These include anxiety disorders, obsessive compulsive disorder, and a strong predisposition to schizophrenia. Schizophrenia develops in about 30% of patients with 22q11DS, making this microdeletion the strongest known link between any genetic anomaly and schizophrenia (Chow and others 2006; Murphy and others 1999; Pulver and others 1994).
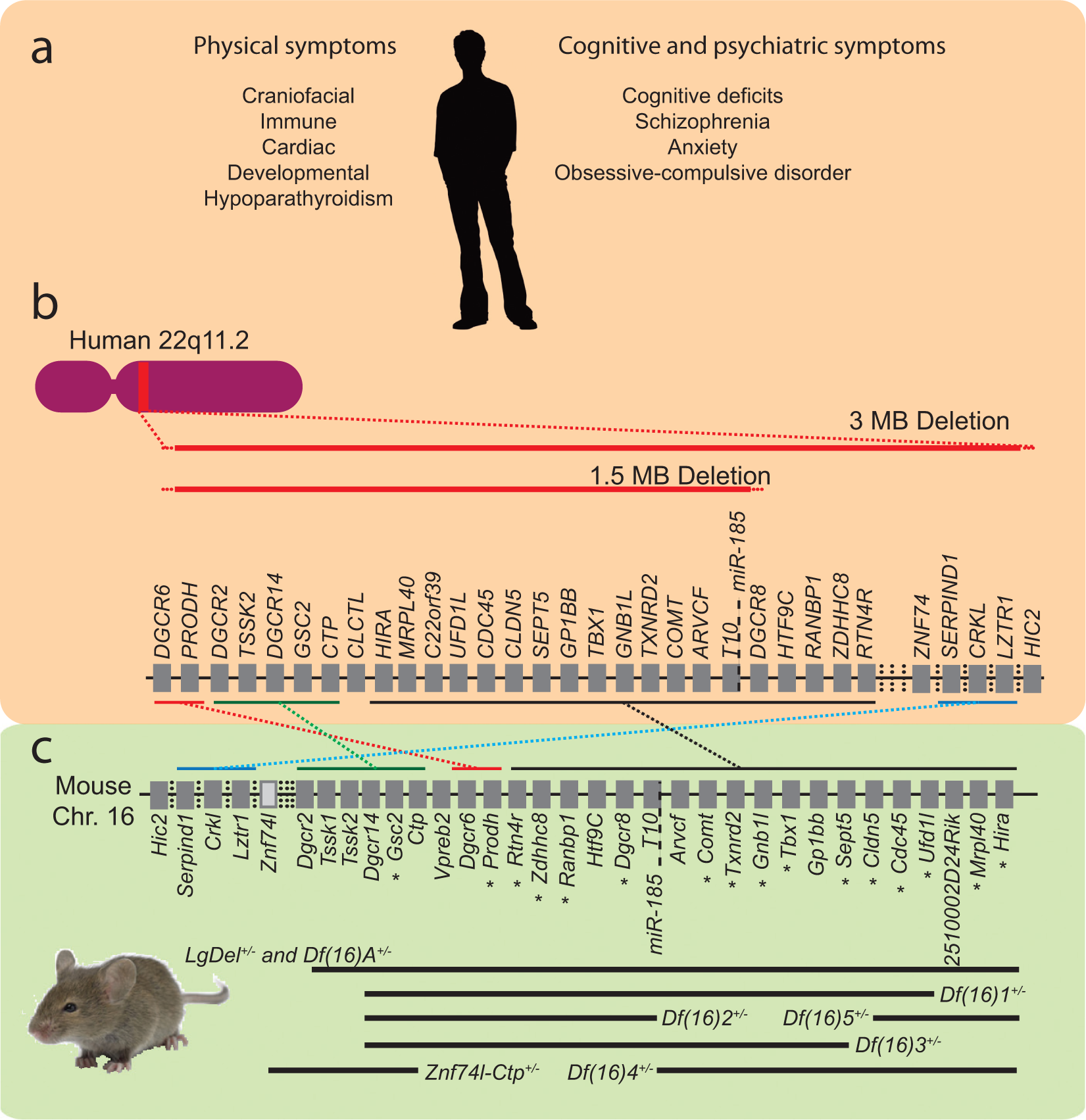
22q11deletion syndrome: modeling human disease in mice. (a) Patients with 22q11DS experience a variety of physical, cognitive, and psychiatric symptoms, some of which are listed. (b) In humans, the same symptoms are caused by either a large (3 MB) or small (1.5 MB) microdeletion of the q arm of chromosome 22. (c) The same group of genes can be deleted from murine chromosome 16 producing various mouse models of 22q11DS. Dark grey rectangles represent individual genes. A light grey rectangle represents a pseudogene. Some genes are not shown (vertical dotted lines). Asterisks represent available mouse strains that carry hemizygous deletions of individual genes within the 22q11-relevant region. Thin horizontal lines show the syntenic regions in humans and mice. Breakpoints of microdeletions vary among patients. Breakpoint-location variability, which is based on Bittel and others (2009), is represented by red dotted lines in (b). Thick horizontal lines show large and small microdeletions in humans (red) and microdeletions of various sizes in available mouse models (black).
The complexity of 22q11DS is as staggering as that of schizophrenia, and the multifaceted psychiatric outcomes associated with 22q11DS indicate that multiple brain circuits are affected. Furthermore, variable penetrance of psychiatric phenotypes suggests a complex interaction of the deletion with other factors such as environmental or epigenetic influences. Clinical features related to the deletion show some overlap with those of associated psychiatric diseases. In schizophrenia, cognitive symptoms (more than positive or negative symptoms) are highly predictive of long-term prognosis (Green 1996). The cognitive disability profile of patients with 22q11DS who develop schizophrenia differs from that of 22q11DS patients that do not develop schizophrenia (Chow and others 2006; van Amelsvoort and others 2004). This suggests either a link between the severity of cognitive symptoms and the development of schizophrenia or an additive effect of the cognitive disability associated with the deletion and a separate form of cognitive disability brought on by schizophrenia.
A multidisciplinary approach is required to understand complex diseases such as 22q11DS and schizophrenia. This review focuses on the study of dysfunctional synaptic connections between neurons in mouse models of 22q11DS and how this approach expands our current understanding of the disease. Such studies are performed in preclinical, genetic models and provide crucial mechanistic information that cannot be obtained in patients. Combining results from these studies with those from clinical studies may help unravel the complex psychiatric outcome associated with the deletion. Thus, because of the strong association between the two disorders, studies of the 22q11DS brain may also provide insight into the synaptic dysfunction associated with schizophrenia.
Challenges to Understanding Complex Psychiatric Disease: The Search for a Meaningful Phenotype
One challenge to studying psychiatric diseases is the lack of tangible readouts. Although some diseases provide specific pathologic markers that give clues about their etiology, deficits in neural function do not always have clear morphologic correlates. Conversely, observed structural changes can be misleading, because they are not always true markers of functional changes. Many current studies rely on observed structural changes as an endpoint, assuming that functional deficits are implied by these morphologic features. Fewer studies go on to determine whether structural alterations contribute to the symptoms of the disease. The result is a body of research that concludes with the assumption that function is compromised if structural changes are present. The impact of these structural findings on brain function (if any) remains to be determined.
For example, postmortem studies of dendritic spine density in the schizophrenia brain have been performed by multiple groups, some of whom have reported dendritic spine density changes associated with the disease. However, it is not clear whether these structural changes are among the causative mechanisms of the disease or whether they are the result of antipsychotic treatment or environmental deprivation associated with institutionalization (Glausier and Lewis 2012). We should exercise caution when interpreting differences in spine number in the diseased brain as representative of changes in neuronal function or as causative.
Similarly, numerous studies in animal models use the density of dendritic spines as a final readout of brain dysfunction. This approach should not be substituted for a direct study of neuronal function for several reasons. First, a typical pyramidal neuron, the principal excitatory neuron in the hippocampus and many areas of the neocortex, has multiple apical, oblique, and basal dendrites, and the dendritic spine density is highly variable between branches, even in a single neuron. Therefore, spine density studies, which tend to focus on a limited region of the dendritic tree, may be prone to overestimation, especially when the comparison involves spine densities of different neuronal populations from animals of different genotypes (Fig. 2a). Second, mild differences in spine counts described in mouse models of psychiatric disease are likely insignificant for function. Typically, mouse models for schizophrenia may show a 10% to 30% change in spine density that is specific to a certain dendritic region (Barros and others 2009; Devito and others 2011; Hayashi-Takagi and others 2010; Kvajo and others 2008; Lee and others 2011; Mukai and others 2008; Xu and others 2013). However, a slight change in only one area of the dendritic tree may account for only a small proportion of overall spines and may have little-to-no effect on overall function (Fig. 2b). Even if spine counts were performed throughout the entire dendritic tree, it would be difficult to estimate the percentage of spine loss/gain that represents functional changes in neuronal output. These confounds may be even more severe in studies conducted in nonintact preparations such as dissociated neuronal cultures, which develop in an artificial state and may be governed by very different developmental and functional rules than a neuron that develops in an intact brain.
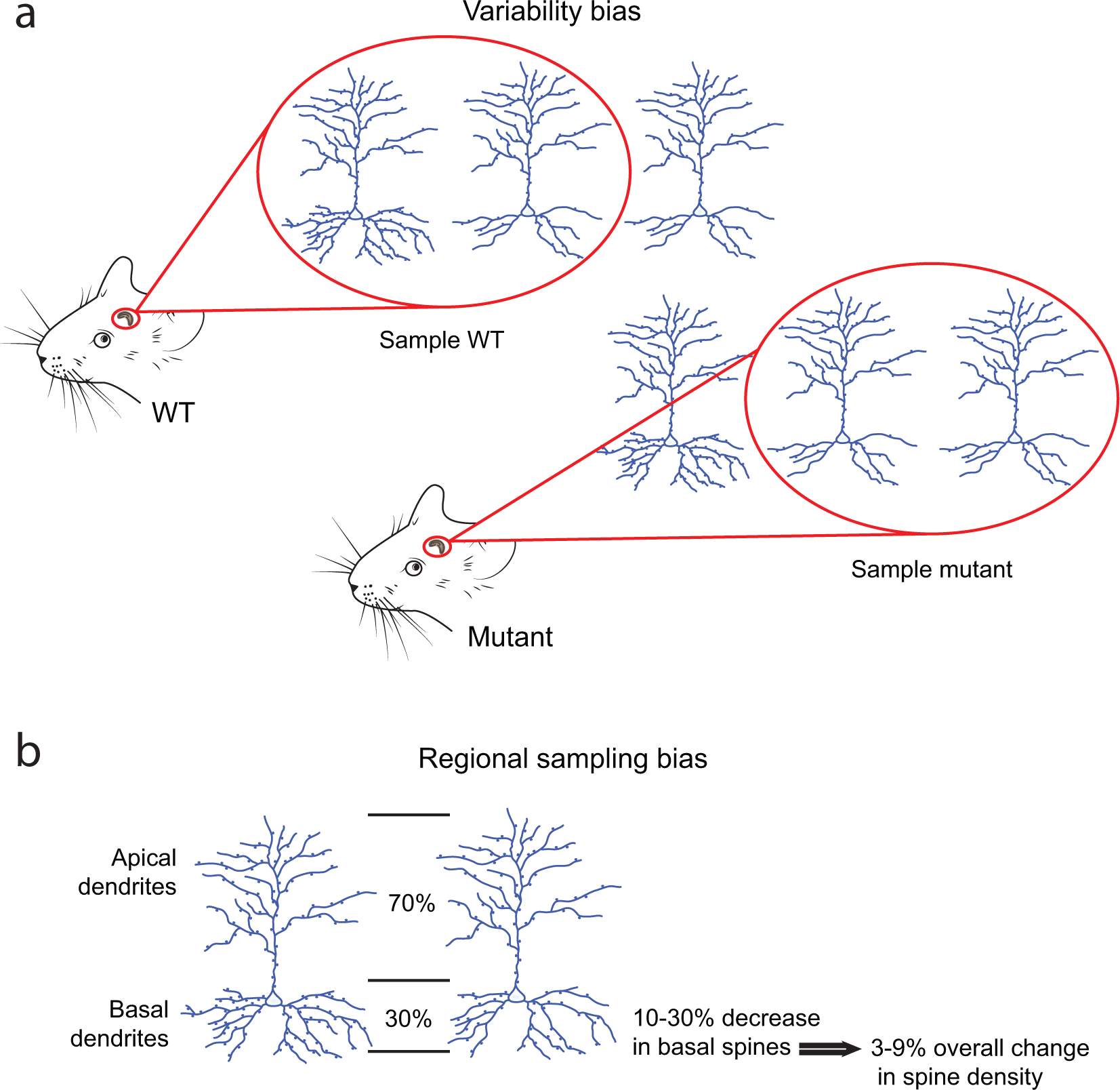
Experimental pitfalls to studying dendritic spine density in models of psychiatric disease. (a) High variability in spine number between neurons and individuals may lead to biased assignment of differences between groups. (b) Potential overestimation of dendritic spine changes identified in a specific region of the neuron. While spine number may change in one region of the dendritic tree, this region may account for a small fraction of total number of dendritic spines, resulting in an overestimation of the spine number difference.
Changes in number of dendritic spines are often ascribed to synaptic plasticity. The idea of “structural synaptic plasticity” is based on numerous observations that dendritic spines change in size and/or number with learning and that these changes can be long lasting (Matsuzaki and others 2004; Yang and Zhou 2009). However, several caveats should be considered when evaluating such claims: First, changes in spine number and size cannot account for the all functional changes observed during synaptic plasticity (Lang and others 2004; Segal 2005). Second, not every dendritic spine undergoes long-lasting size changes; in juveniles and adults, most dendritic spines are stable (Grutzendler and others 2002) and undergo only transient structural changes during synaptic plasticity (Lang and others 2004). Many studies in models of schizophrenia are conducted at earlier ages, when spines are more dynamic. This increases the probability of finding differences that may be irrelevant to disease symptoms, which become apparent at later ages. For example, spine differences observed at young ages may be the result of developmental delay and may resolve by adulthood, disappearing by the time of disease onset. Third, all dendritic spines are not created equal. Spines occupying different locations on dendrites and having different structural and biochemical properties may contribute differently to the postsynaptic response (Richardson and others 2009). Synapses associated with some spines are silent, because they lack postsynaptic receptors or have a silent or immature presynaptic terminal (Crawford and Mennerick 2012; Kerchner and Nicoll 2008). Thus, studies that involve counting spines as a final readout assay have no way of identifying whether those spines are active and capable of contributing to neuron function.
The generation of an action potential requires the summation of only a few out of the 10,000 synaptic inputs of a typical pyramidal neuron (McNaughton and others 1981; Otmakhov and others 1993). Is it therefore likely that minor changes in spine number affect action potential generation? The answer may depend on what types of spines are gained or lost and their locations on the dendrite. It is easy to envision that the loss of a few dendritic spines on a vast dendritic tree would not affect the output of a pyramidal neuron (Fig. 3). Therefore, structural abnormalities with no functional correlates may lead to more confusion rather than genuine understanding of these diseases.
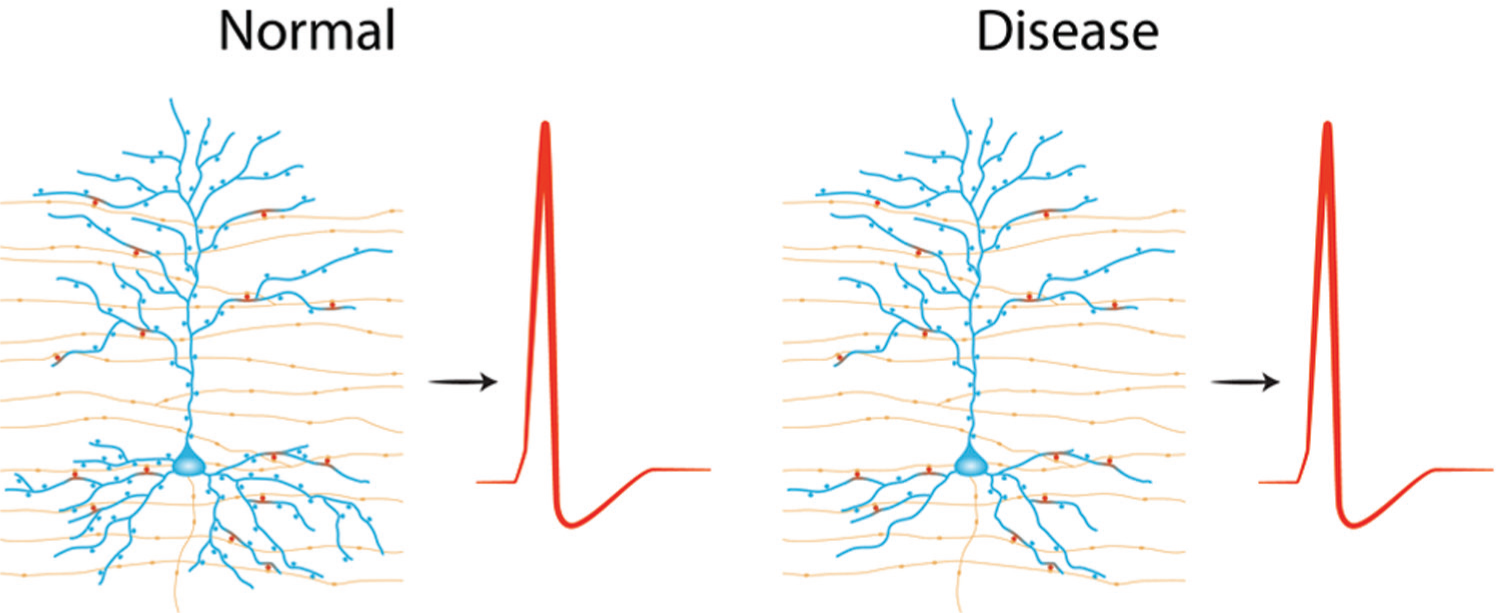
Dendritic spine density is not an accurate predictor of neuronal function. The depicted scenario illustrates how the elimination of a few dendrites and dendritic spines by disease may not necessarily affect the generation of action potentials in postsynaptic neurons. Yellow indicates axons with presynaptic boutons; blue, postsynaptic neurons with dendrites and dendritic spines; and red, activated dendritic spines.
Moreover, structural abnormalities can be secondary to functional deficits rather than causative. This is illustrated by a recent magnetic resonance imaging study of the schizophrenia brain. In this study, hippocampal function and structure were compared in a cohort of patients over time. The authors found that functional disturbances precede and lead to hippocampal atrophy in schizophrenia rather than atrophy causing the disturbances. The study further suggested that an abnormally high level of glutamate release is the primary driver of this psychotic disorder rather than structural abnormalities (Schobel and others 2013). Thus, the search for mechanisms responsible for abnormally high levels of glutamate release, rather than neuronal atrophy or a decrease in the density of dendritic spines in the hippocampus, may be a more informative approach to identifying the pathophysiologic mechanisms of schizophrenia.
Genomic profiling may also not be an ideal means to study schizophrenia because of the complexity of the disease. Schizophrenia symptoms most likely arise from malfunctions of multiple circuits in several brain regions (Lisman and others 2010; Sigurdsson and others 2010). The deficits in each circuit are not necessarily the same; they may originate from diverse molecular or anatomic defects. This level of complexity can impede our understanding of disease by common reductionist approaches. Consider a disease in which gene expression is affected differentially in different neuronal subtypes. A typical genomic-profiling experiment of a brain region composed of multiple neuronal subtypes may not detect subtle variations in specific cell subtypes. For example, a relevant expression change that is specific to an interneuron that constitutes 1% of overall cells in a brain region would likely go undetected in a profile of the entire tissue. Furthermore, genomic approaches often focus on specific brain regions in isolation and do not detect the additive effects of different genes malfunctioning in different but interconnected regions of the brain. This type of complexity is either undetectable or easily misinterpreted by a genomics approach. Examining brain malfunction in psychiatric disease at the circuit level may identify molecular pathways that are beyond the grasp of genomics studies.
Although mounting evidence points to synaptic dysfunction in schizophrenia, only a few synaptic studies have been done. Differences in dendritic morphology and gene expression may imply functional differences, but they may also be inconsequential to neuronal function, epiphenomena of the disease, or a consequence of drug treatments. For diseases with valid preclinical model systems, we can directly assess synaptic function of different circuits. Thus, functional assays can be established as readouts of contributing molecular and/or morphologic mechanisms. A synaptic function approach casts a broader net than pure molecular approaches, yet is more mechanistically specific and more sensitive than approaches such as behavioral assays or low-resolution brain imaging.
Mouse Models of 22q11DS
Although directly studying the human condition is ideal, genetic variability, pharmacologic intervention, and broad environmental influences confound human research. Studies in preclinical genetic models are less encumbered by these variables, as genetic and environmental variability are minimal, and pharmacologic intervention can be controlled. Using a model is particularly advantageous when studying antipsychotics, because these drugs have a broad range of side effects, and patients rarely take them according to the prescribed schedule. Finally, the use of animal models enables researchers to conduct mechanistic studies that would be too invasive to perform in humans.
Excellent mouse models of 22q11DS have been generated in the past 20 years. Most (90%) patients with 22q11DS have a 3-MB deletion of approximately 40 genes in the 22q11.2 locus; others have a 1.5-MB deletion within the same locus (Fig. 1b). The clinical manifestation of the 1.5-MB deletion is indistinguishable from that of the 3-MB deletion (Carlson and others 1997). Therefore, the disease-causing genes are probably contained within the 1.5-MB region. Fortunately for researchers, this region is conserved on mouse chromosome 16 (Fig. 1c). Mouse models of the 1.5-MB deletion carry a heterozygous deletion of a large region encompassing most of the disease-critical genes. Several such models have been reported: two carrying a deletion between the Dgcr2 and Hira genes (referred to as Lgdel+/− mice [Merscher and others 2001] and Df(16)A+/− mice [Stark and others 2008]) and another carrying a deletion between the Dgcr14 and Ufd1L genes [Df(16)1+/− mice; Lindsay and others 1999]. Additionally, mice with smaller deletions within the syntenic region of chromosome 16 have been generated (Kimber and others 1999; Lindsay and others 2001; Puech and others 2000). These strains allow for the narrowing of genetic regions responsible for specific phenotypes of 22q11DS (Paylor and Lindsay 2006). Finally, mouse lines carrying single mutations of nearly all of the genes within this region have been generated, including Gsc2 (Saint-Jore and others 1998), Prodh (Gogos and others 1999), Zdhhc8 (Earls and others 2012; Mukai and others 2008), Ranbp1 (Nagai and others 2011), Dgcr8 (Earls and others 2012; Hsu and others 2012; Stark and others 2008; Wang and others 2007), Comt (Gogos and others 1998), Txnrd2 (Kim and others 2004), Gnb1l, Tbx1 (Lindsay and others 2001), Sept5 (Peng and others 2002), Cldn5 (Nitta and others 2003), Cdc45l (unpublished), Ufd1l (Lindsay and others 1999), Mrpl40, and Hira (Roberts and others 2002). Once mutant strains of every gene within the syntenic region are available, we will be able to conduct full coverage screens for individual genes responsible for phenotypes discovered in the large deletion models. The collection of these models can be a powerful genetic tool to probe neuronal function and synaptic connections throughout the brain.
Because schizophrenia is a common consequence of the 22q11 deletion, mouse models of 22q11DS are becoming synonymous with models of schizophrenia. However, considering the overlap of their clinical features, we must be cautious equating these two diseases and assuming that results from mouse models of 22q11DS can be generalized to schizophrenia. For example, both diseases are associated with defects in social interaction. Schizophrenia patients often show a lack of emotional responsiveness and become socially withdrawn. Patients with 22q11DS also have a high rate of social symptoms, due at least in part to social anxiety. The result (i.e., reduced social interaction) is the same, but the underlying causes may be quite different. If social deficits are described in a model system of 22q11DS, do we attribute them to 22q11DS or schizophrenia? How do we know that a particular phenotype is related to 22q11DS, schizophrenia, or common to both diseases? Such questions can be answered by pursuing hypotheses formed from mouse model experiments in patient populations.
One would predict that schizophrenia-specific mechanisms are present in schizophrenic patients, whether or not they carry the 22q11.2 deletion. Conversely, 22q11DS-specific mechanisms would be present in patients carrying the deletion, regardless of whether they have schizophrenia, but not in the general schizophrenia population. Clinical follow-up is vital to validate these models and elucidate the causes of different phenotypes of 22q11DS. One example of a mouse model discovery that translates to human disease is the upregulation of the sarco(endo)plasmic reticulum Ca2+ ATPase type 2 (SERCA2), found in the brains of both mouse models of 22q11DS and human schizophrenia patients (Earls and others 2010; Earls and others 2012). SERCA2 upregulation in the general schizophrenia population suggests that this is one molecular change that may be important for cognitive aspects of schizophrenia rather than specific to 22q11DS cognitive symptoms. Studies in both models and patients are especially important for brain regions affected by both diseases. For example, the hippocampus is strongly implicated in the cognitive deficits of both diseases, whereas phenotypes described in the sensory cortices may be more easily generalized to psychosis.
It is not unreasonable to predict that findings of synaptic or neuronal dysfunction in mouse models have great potential to help resolve the complex neural pathogenesis of both diseases. However, directly drawing conclusions about schizophrenia from 22q11DS mouse models without follow-up studies in patients may only confuse both fields.
A Synaptic Plasticity Approach to Understanding Psychiatric Disease
Many molecular and cellular processes must come together to create a functioning synapse. Normal communication between two neurons requires the propagation of action potentials along the axon of a presynaptic cell, probabilistic release of neurotransmitter from synaptic vesicles at presynaptic boutons, and activation of postsynaptic receptors that, in turn, leads to cascades of electrical and molecular events in a postsynaptic neuron. Synaptic communication, however, is not merely a matter of transmission between neurons. Synaptic transmission can be altered by prior activity allowing for the storage of information at synapses. Synaptic plasticity, an activity-dependent bidirectional change in synaptic strength, is present in most excitatory and inhibitory synapses throughout the brain. Multiple mechanisms induce and maintain synaptic plasticity, which comprises several components even at a single synapse (Blundon and Zakharenko 2008; Citri and Malenka 2008; Lisman and Raghavachari 2006).
Synaptic plasticity is believed to be the cellular mechanism of cognition (Martin and others 2000; Milner and others 1998; Whitlock and others 2006), which is progressively affected in both patients with 22q11DS (Gothelf and others 2007; Green and others 2009) and schizophrenia (Keefe and Harvey 2012). Therefore, a detailed understanding of the molecular pathways governing synaptic plasticity can help us better understand the cognitive symptoms of psychiatric disease. Because synaptic plasticity is a complex phenomenon that involves multiple molecular events in presynaptic and postsynaptic neurons, disease-associated changes in synaptic plasticity can result from many potential cellular insults. Synaptic plasticity can also be affected indirectly, even if the core plasticity mechanisms remain intact. For instance, if neuronal excitability or basal synaptic transmission is compromised, then synaptic plasticity may also be affected. Because of this, synaptic plasticity studies provide the ultimate functional readout of a given neuronal circuit and report the net effect of disease or a disease-associated mutation in the context of that circuit. Conversely, cellular changes that are functionally inconsequential are filtered out in such studies. For example, a disease may cause the loss of a few dendritic spines (Fig. 3) or change gene expression. If the cell compensates, or if those changes do not ultimately affect neuronal or synaptic function, then their irrelevance will be detected in a study of synaptic function. Whereas a genomics or morphologic study might identify these changes and incorrectly implicate them in the disease, a synaptic function approach would only identify changes that functionally affect the circuit studied.
Synaptic function is highly sensitive to even minor changes in relevant gene expression or function. For example, very small changes in the proteins that maintain Ca2+ levels can create detectable changes in synaptic vesicle release (Heidelberger and others 1994). As a consequence, mutations that cause minor changes in Ca2+ entry into the cell profoundly affect synaptic plasticity. For the purpose of understanding a complex psychiatric disease, the synaptic function approach can complement other methodologies by providing a sensitive functional readout of malfunctioning neuronal circuits.
Hippocampal Synaptic Plasticity Is Altered in 22q11DS Mice in an Age-Dependent Manner
Recent findings in the Df(16)1+/− mouse illustrate how a synaptic function approach provides an informed basis for subsequent molecular experiments (Earls and others 2010; Earls and others 2012). The first question to ask when designing a synaptic function study is which neuronal circuits are the most critical for disease symptoms? Multiple brain regions have been associated with the symptoms of 22q11DS and schizophrenia. Because cognitive symptoms are central in both conditions and are not amenable to pharmacologic therapies, synapses implicated in cognitive symptoms are among the most essential to characterize. The hippocampus is a region of the brain that is crucial for learning and memory. Alterations in the hippocampus have been observed in patients with 22q11DS and/or schizophrenia (Chow and others 2002; Debbane and others 2006; Deboer and others 2007; Heckers and others 1998; Medoff and others 2001).
Because of the likelihood of hippocampal involvement in 22q11DS and schizophrenia, our laboratory recently characterized the function of excitatory synapses between CA3 and CA1 pyramidal neurons (CA3-CA1 synapses) of the hippocampus in the Df(16)1+/− mouse model of 22q11DS. Basal synaptic transmission and short- and long-term synaptic plasticity (in the form of long-term potentiation or LTP) were unaltered in young (~8-week-old) animals carrying the deletion (Earls and others 2010). However, because the age of schizophrenia onset is 18 to 35 years in humans, we reasoned that synaptic deficits in the hippocampus of this model might be detectable in more mature animals. Indeed, we found that short- and long-term synaptic plasticity are dramatically enhanced in older (~16-week-old) Df(16)1+/− mice (Earls and others 2010). This synaptic plasticity phenotype was highly robust and constituted a more than 100% increase in short- and long-term potentiation of synaptic transmission compared to that seen in wild-type mice. Synaptic transmission, on the other hand, was unchanged in older Df(16)1+/− mice, suggesting that basal synaptic properties (i.e., connectivity of CA3 and CA1 neurons, morphology of the pyramidal neurons, release of glutamate from synaptic vesicles in presynaptic terminals, number of postsynaptic glutamate receptors, and other properties of synaptic transmission) do not change in animals that carry the 22q11-related microdeletion. These findings demonstrate that the 22q11 microdeletion specifically affects mechanisms of synaptic plasticity and does so in an age-dependent manner.
It is important to distinguish synaptic transmission from synaptic plasticity, because sometimes this distinction is obscured in studies of psychiatric disease. Any relevant changes in dendritic morphology or density of dendritic spines should affect neuronal connectivity and, as a consequence, basal synaptic transmission. Because all reports so far indicate that synaptic transmission is normal in young and mature 22q11DS mice (Drew and others 2011; Earls and others 2010), the morphology of hippocampal neurons probably is either unaffected by the microdeletion (Earls and others 2010), or its effect is inconsequential for synaptic transmission. It may therefore be preferential to study the cognitive symptoms 22q11DS microdeletion from the point of view of synaptic plasticity, which has long been correlated with learning and memory (Martin and others 2000; Milner and others 1998; Whitlock and others 2006), and which provides a more definitive endpoint for scientific study.
Cellular Mechanisms of Abnormal Synaptic Plasticity in 22q11DS
Synaptic plasticity at excitatory synapses can involve presynaptic and postsynaptic changes (Bayazitov and others 2007; Blundon and Zakharenko 2008) and are modulated by an ever-growing number of molecular pathways (Sanes and Lichtman 1999). Therefore, to determine the mechanism of synaptic plasticity changes in Df(16)1+/− mice, we first narrowed the cellular locus of synaptic dysfunction. During the last decade, powerful new tools for dissecting presynaptic and postsynaptic function have emerged. Two-photon laser-scanning microscopy (TPLSM) of fluorescently labeled neurons visualizes fine subcellular structures, including presynaptic terminals and postsynaptic dendritic spines. TPLSM of the hippocampus can be performed in acute brain slices in which functional circuitry is maintained, and detailed imaging and electrophysiological experimentation are feasible. This preparation allows neurons to develop and form proper functional connections in an intact brain. Therefore, slice preparations are more biologically relevant in the context of neuronal circuits than more reduced preparations such as dissociated cultured neurons.
Fluorescent dyes can be introduced inside an individual neuron through a miniature glass pipette that is typically used for electrophysiological recordings. This approach allows for labeling of a cell of interest and visualization of its entire dendritic structure with high resolution. We used this approach to test whether the 22q11-related deletion affects the morphology of CA1 pyramidal neurons in the hippocampus of Df(16)1+/− mice (Earls and others 2010). Because the majority (~64%) of CA3-CA1 excitatory synapses is located on apical dendrites, we compared the overall dendritic structure and dendritic spines of apical dendrites. We observed no differences in the dendritic spine size, density, or type and no difference in the branching of apical dendrites in young or mature Df1(16)1+/− mice and wild-type littermates (Earls and others 2010). Thus, structural or synaptic changes in those neurons most likely do not underlie synaptic plasticity abnormalities in this model. This finding is consistent with normal basal synaptic transmission observed at CA3-CA1 synapses of 22q11DS mice (Drew and others 2011; Earls and others 2010) and implies that functional rather than structural mechanisms influence synaptic plasticity in these mice.
Changes in synaptic plasticity at CA3-CA1 synapses are compound in nature, that is, they can be the result of presynaptic deficits, postsynaptic deficits, or both (Bayazitov and others 2007; Blundon and Zakharenko 2008). Currently, limited tools are available to identify the cellular locus of synaptic abnormalities. Two-photon glutamate uncaging specifically probes postsynaptic function by releasing exogenous glutamate on a single dendritic spine (Fig. 4a) (Bloodgood and Sabatini 2005; Matsuzaki and others 2004). “Caged” (inert) glutamate in the extracellular space becomes active (uncaged) on illumination by light of certain wavelengths. Two-photon excitation removes a “cage” in a volume as small as a femtoliter, providing spatial and temporal precision of glutamate delivery equivalent to the release of endogenous glutamate from a presynaptic terminal. By controlling the duration, intensity, and frequency of two-photon illuminations, one can deliver glutamate to individual dendritic spines with similar kinetics and frequencies as those of endogenous glutamate released from a presynaptic terminal. By circumventing the presynaptic terminal, we can study postsynaptic function in isolation. Using this approach, we found no deficiency in the postsynaptic component of synaptic plasticity in Df(16)1+/− mice (Earls and others 2010). We therefore hypothesized that synaptic plasticity abnormalities in 22q11DS arise from presynaptic deficiencies.
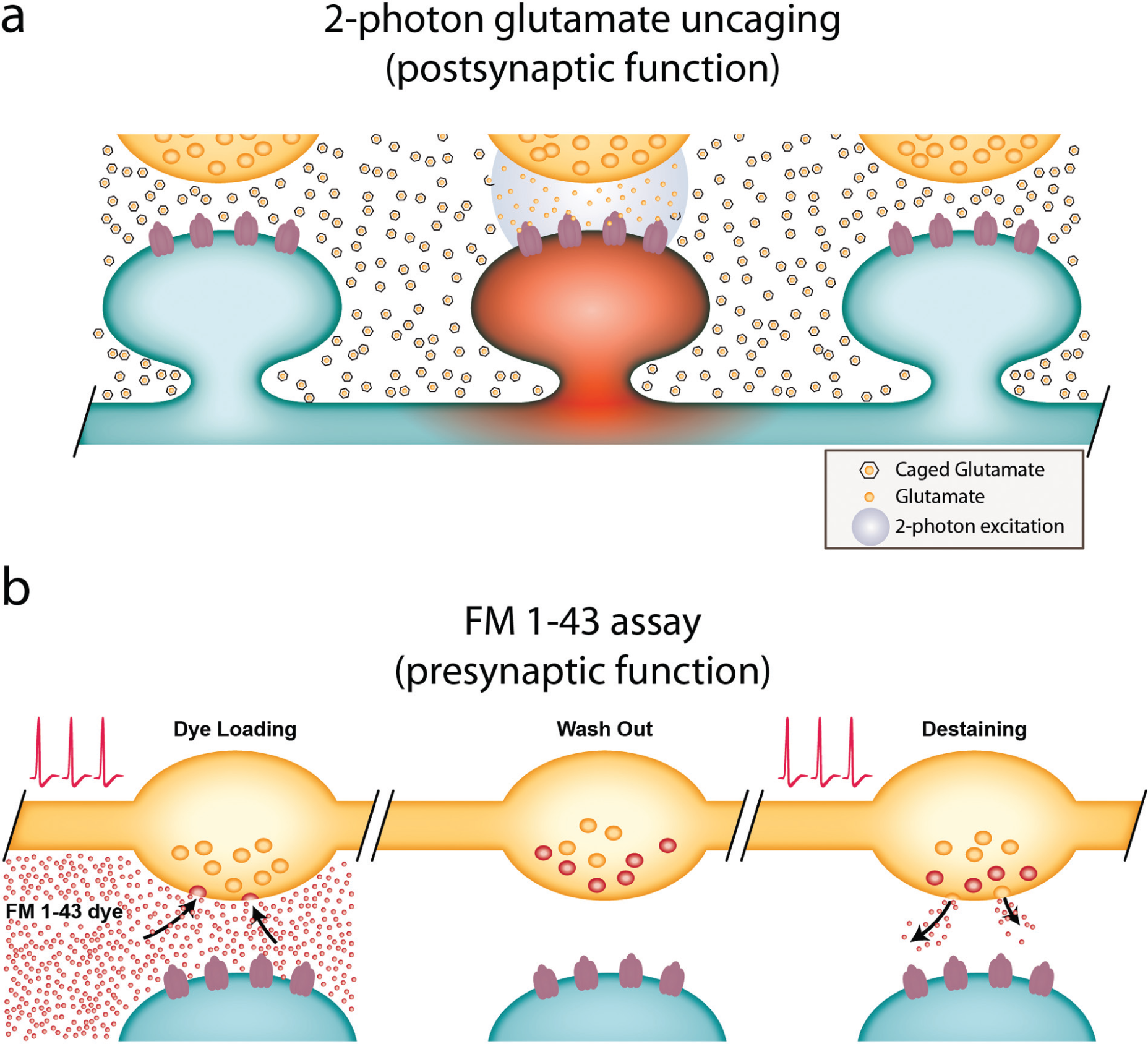
Direct methods to measure postsynaptic and presynaptic function at individual synapses. (a) Two-photon glutamate uncaging directly assesses postsynaptic function of individual dendritic spines. Exogenous caged (inert) glutamate is activated by two-photon light excitation of certain wavelengths. Once uncaged (active), the exogenous glutamate acts on the postsynaptic dendritic spine, as would glutamate released from the presynaptic terminal. (b) The FM 1-43 assay measures release from presynaptic boutons. FM 1-43 is applied to brain tissue and the dye is taken up into recycling synaptic vesicles (dye loading) after stimulation. The remaining exogenous dye is then removed (wash out), and the cell is electrically stimulated again. The amount of FM 1-43 released (destaining) from the presynaptic terminal is measured as a proxy for neurotransmitter release from synaptic vesicles. Small dots represent FM 1-43 dye.
This theory was confirmed by several lines of evidence using imaging tools specific for testing presynaptic function. One such tool is the FM 1-43 assay, which can be applied in various neuronal preparations including acute brain slices (Kay and others 1999; Zakharenko and others 2003). This method exploits the fact that FM 1-43 dye (Betz and Bewick 1992) is taken up into synaptic vesicles during activity-dependent endocytosis (Fig. 4b). FM 1-43 is not fluorescent in water-based solutions but becomes fluorescent when bound to lipids. This allows not only for the detection of active presynaptic terminals but also for the quantitative analysis of activity-dependent release of FM 1-43 dye from labeled synaptic vesicles, which is then used as a measure of neurotransmitter release. Compared to other methods for testing presynaptic activity, which mostly rely on indirect electrophysiological analysis of excitatory postsynaptic currents, the FM 1-43 assay directly measures presynaptic function. Another advantage of this assay is that it can measure presynaptic activity at individual presynaptic terminals, whereas electrophysiological approaches test the net effect of all inputs to a given neuron.
Using the FM 1-43 assay in acute hippocampal slices, we and others have shown that synaptic plasticity at CA3-CA1 synapses has a substantial presynaptic component (Stanton and others 2001; Zakharenko and others 2001; Zakharenko and others 2002; Zakharenko and others 2003). We used this assay to test presynaptic function at CA3-CA1 synapses of 22q11DS model mice. We found accelerated FM 1-43 release from presynaptic terminals of Df(16)1+/− mice during plasticity induction (Earls and others 2010), further indicating that presynaptic mechanisms of synaptic plasticity underlie the age-dependent increase in LTP in these mice.
Ca2+ is a key regulator of neurotransmitter release, and because of the highly cooperative relationship between presynaptic Ca2+ and neurotransmitter release (Borst and Sakmann 1996), even a slight perturbation in presynaptic Ca2+ regulation can profoundly affect neurotransmitter release. Because Ca2+ is so critical for transmitter release, we used TPLSM to measure Ca2+ dynamics in individual presynaptic terminals of CA3 hippocampal neurons in Df(16)1+/− mice (Fig. 5). We loaded CA3 pyramidal neurons with a fluorescent Ca2+ indicator and a cytoplasmic fluorescent dye through the recording pipette (Fig. 5a). This approach allowed us to visualize the dendrites and axon with TPLSM (Fig. 5b) and to deliver a desired number of action potentials through the recording pipette. We identified presynaptic boutons on CA3 axons and measured Ca2+ transients evoked by a train of presynaptic activity. We observed an increase in Ca2+ entering the presynaptic terminal on stimulation and a decrease in the rate of decay of Ca2+ transients in Df(16)1+/− mice compared to wild-type littermates (Earls and others 2010) (Fig. 5c). This result suggested that the 22q11deletion results in dysregulation of presynaptic Ca2+. Further experiments suggested that the microdeletion affects endoplasmic reticulum uptake, storage, or release of Ca2+ from the endoplasmic reticulum.
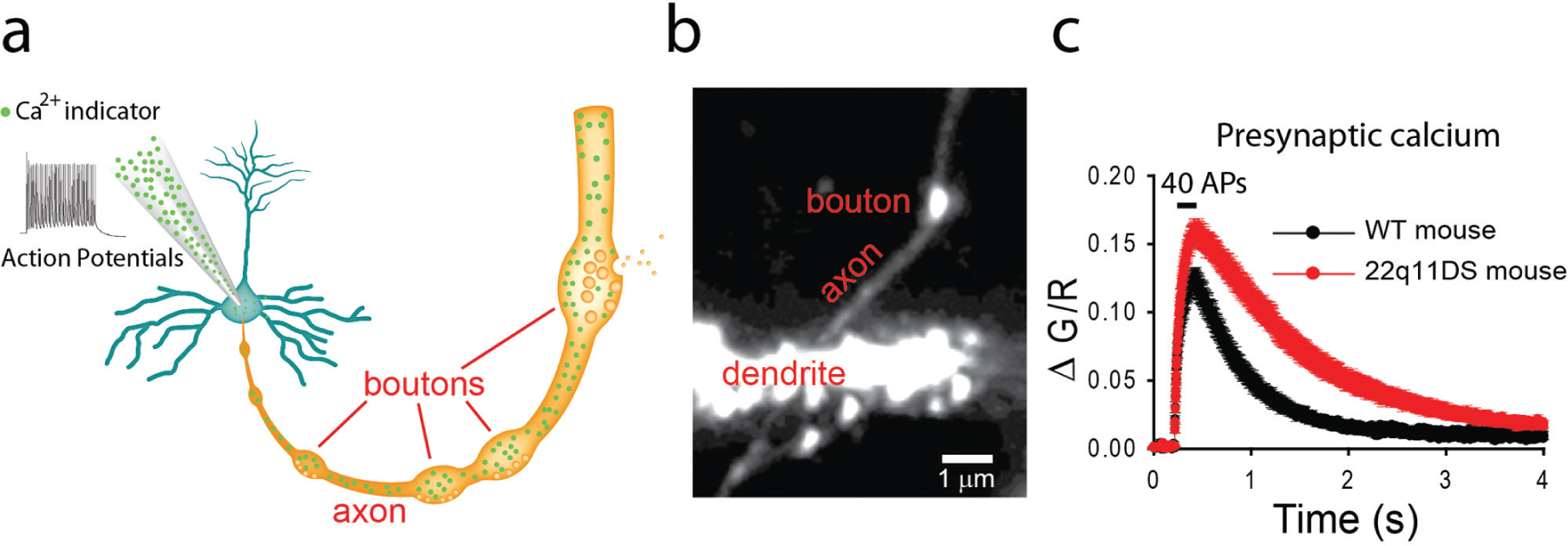
Abnormal calcium dynamics in presynaptic boutons of 22q11DS models. (a) Paradigm for measurement of presynaptic Ca2+ dynamics. A presynaptic CA3 neuron is connected to a glass pipette filled with a cytoplasmic fluorescent dye (not shown) and a Ca2+ indicator (small green dots) that increases in fluorescence in the presence of Ca2+. The pipette is also used to activate the presynaptic cell body by delivery of a train of action potentials (APs). Ca2+ entry into presynaptic boutons is then measured by TPLSM detection of fluorescent changes of a Ca2+ indicator dye in response to stimulation. (b) TPLSM image of fragments of a dendrite with dendritic spines and an axon with a presynaptic bouton of a CA3 neuron. (c) Presynaptic Ca2+ transients in mouse models of 22q11DS (red) are substantially different than those in wild-type (WT) littermates (black). Values are reported as ΔG/R, the change in Ca2+ indicator fluorescence (G) normalized to change in cytoplasmic dye fluorescence (R).
From Synaptic Plasticity Abnormalities to Molecular Mechanisms of 22q11DS
Using a functional approach, we were able to identify previously unknown cellular mechanisms of abnormal synaptic plasticity in models of 22q11DS. Using this approach put us in a unique position to study hippocampal dysfunction at the molecular level in 22q11DS. First, functional studies revealed that presynaptic Ca2+ is dysregulated in 22q11DS mice, prompting us to focus on proteins that regulate Ca2+ inside the synapse. Serca2, which pumps Ca2+ into the endoplasmic reticulum, is increased in the brains of 22q11DS mice (Earls and others 2010). This increase is brain-specific and observed only at the protein level, not the transcript level, suggesting posttranscriptional dysregulation of Serca2 in 22q11DS. Elevated Serca2 was subsequently shown to dysregulate presynaptic Ca2+ and LTP in Df(16)1+/− mice (Earls and others 2010).
Second, ~100% enhancement in LTP in 22q11DS model mice is a highly robust phenotype. Whereas subtle phenotypes of behavior or morphology may show variable reproducibility between genetic strains, strong phenotypes such as this increase in LTP are reproducible enough to be used to screen various mutant mouse strains for molecular pathways that contribute to disease. Using the LTP increase as a readout, we screened the existing mouse strains that have hemizygous deletions of small clusters or individual genes within the deletion region. This screen identified Dgcr8 as a culprit gene that contributes to the age-dependent LTP increase (Earls and others 2012). Further study showed that the hemizygous loss of Dgcr8 is responsible for Serca2 upregulation. Dgcr8 is a double-stranded RNA-binding protein important for microRNA (miRNA) biogenesis (Tomari and Zamore 2005), and thus its haploinsuffuciency has the potential to affect translation of miRNA targets. We therefore hypothesized that the 22q11 microdeletion depletes miRNAs that target the Serca2 mRNA.
To test this, we performed microarray analysis to determine whether miRNAs that target the Serca2 transcript are affected by the Df(16)1 microdeletion. We found that miR-185 and miR-25 are depleted in these mice, target Serca2 expression, and thus regulate hippocampal LTP, in the Df(16)1+/− mice (Earls and others 2012). Therefore, a functional approach helped us determine that the 22q11 deletion leads to a reduction in Dgcr8, which results in the depletion of key miRNAs that would normally keep Serca2 levels in check. With the depletion of these miRNAs, Serca2 rises to abnormal levels, resulting in dysregulated Ca2+ turnover in presynaptic terminals and abnormally high levels of glutamate release during the sustained neuronal activity that is required for synaptic plasticity at excitatory synapses. Figure 6 illustrates this model for the mechanism of hippocampal dysfunction in 22q11DS.
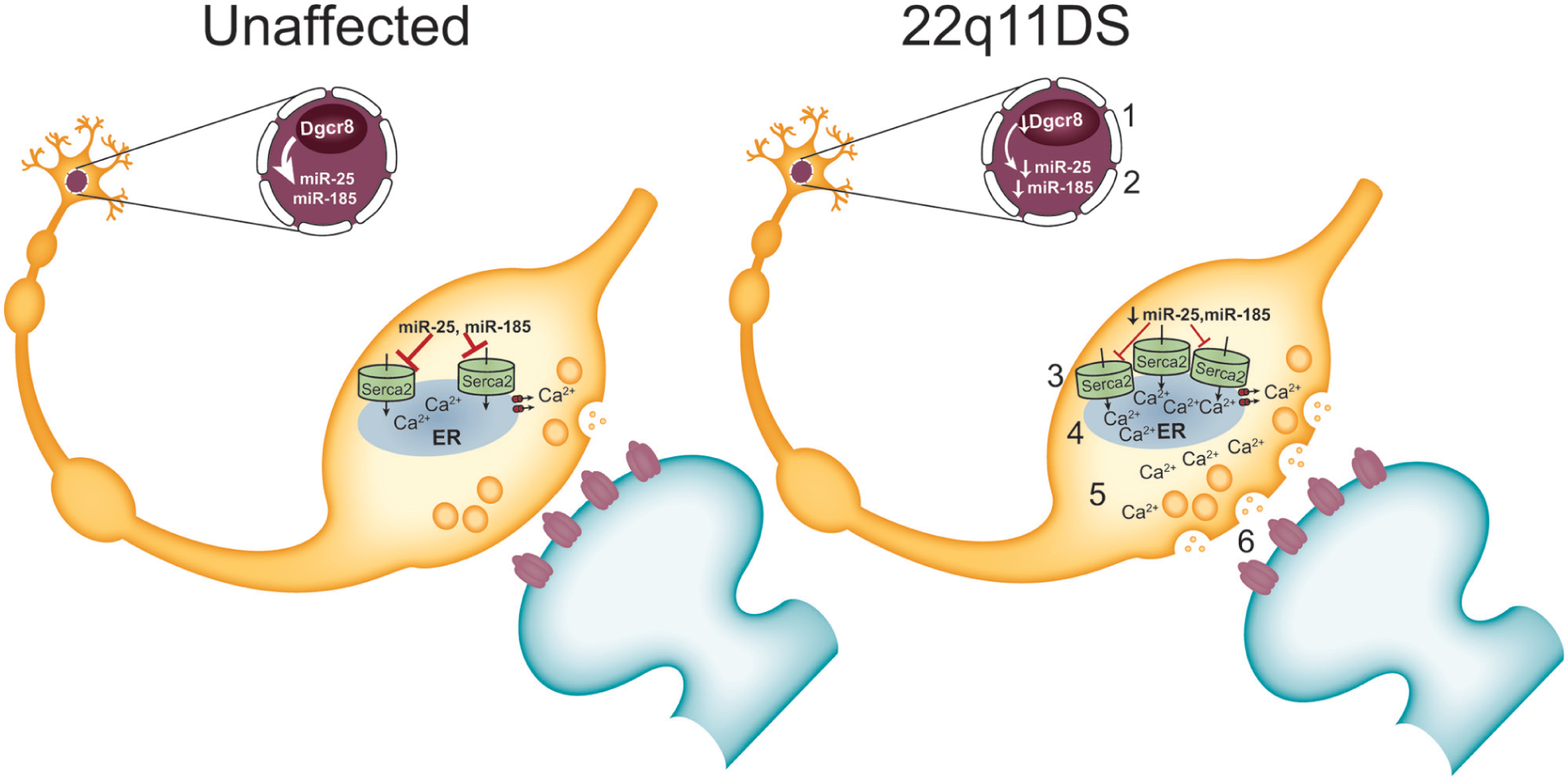
A model of 22q11DS. Results from mouse model studies of 22q11DS suggest the following model for the microdeletion’s effect on hippocampal function. Dgcr8 haploinsufficiency (1) causes a reduction in Serca2-regulating microRNAs miR-185 and miR-25 (2) in 22q11DS. The resultant increase in Serca2 protein (3) causes abnormal Ca2+ build-up in the endoplasmic reticulum (ER) of presynaptic terminals (4). This results in increased Ca2+ release from the ER with neural activity (5) and enhanced glutamate release (6). The overall consequence of this synaptic dysfunction is abnormal synaptic plasticity, which results in impaired learning and memory.
Potential Impact of Functional Approaches on the Study of Psychiatric Disease
When faced with a disease of complex or unknown etiology, traditional high-throughput approaches are often applied as the initial step to understanding the disease. However, these approaches may fail to provide insight or may lead to years of fruitless research on deceptive target genes or phenotypes. Here we present the advantages of the synaptic function approach of first determining the functional effects of disease. Observed functional abnormalities can then be used to guide subsequent molecular studies. This approach has the potential to identify disease molecules missed by strictly molecular approaches. For instance, in 22q11DS mice, Serca2 transcript levels are unaffected, but Serca2 protein levels are increased. This result is undetectable by genomic strategies, and the proteomics field has not yet acquired adequate sensitivity or coverage to have reliably detected Serca2 elevation. This result illustrates how functional experiments followed by molecular studies can identify critical molecular players that have eluded traditional high-throughput molecular approaches.
Next, these results demonstrate that the mechanisms controlling synaptic plasticity, even at a single synapse, are diverse and dynamic throughout a lifetime. Our findings in 22q11DS model mice identified a disease pathway that shows age-dependence (Earls and others 2010; Earls and others 2012). Although the deletion is present in all cells and at all ages, its effects differ, depending on the cellular context and developmental stage of the organism. In the case of 22q11DS model mice, hippocampal dysfunction only becomes apparent at 16 weeks of age, despite the fact that Dgcr8 is depleted throughout life. This emphasizes the age progression of 22q11DS and the importance of studying models of the disease at varying ages. Although early development has been the focus of much research for this disease, crucial disease events, such as Ca2+ dysregulation in the hippocampus, may not occur until adolescence or adulthood. Further research is needed to address the question of why the brain only becomes vulnerable to specific psychiatric diseases at particular ages. This is a central question in neuroscience, but is difficult to address until age-dependent disease events are identified. Further study of the mechanisms of age-dependence of the Dgcr8-Serca2 pathway should give insight into this essential question.
Finally, these studies illustrate the importance of integrating findings from the synaptic function approach into subsequent clinical studies. Our studies in 22q11DS mouse models (Earls and others 2012) proved relevant to schizophrenia in humans. The increase in Serca2 protein levels observed in 22q11DS mouse models was also found in postmortem brain samples from schizophrenic patients (Earls and others 2012). This is an indicator that a pathogenic event caused by the 22q11 deletion can be generalized to schizophrenia. This discovery raises important questions: Is the increase in SERCA2 protein observed in schizophrenia patients the result of depleted miRNAs, or are there alternative mechanisms that modulate SERCA2? A study of SERCA2 regulation in schizophrenia is needed to address this question. Could changes in other cellular pathways result in the same synaptic and neuronal phenotypes as Serca2 upregulation? It is intriguing to speculate that other schizophrenia-associated mutations outside the 22q11 region could result in similar functional outcomes.
Here, we describe synaptic malfunction at a particular synapse. However, a similar approach can be applied to other neuronal circuits that are affected in 22q11DS and schizophrenia. Because brain function is the ultimate target of psychiatric disease, we advocate approaching these diseases by identifying functional rather than structural abnormalities in neurons and synapses in model organisms. Further investigation of mechanisms of such functional abnormalities will determine the underlying cellular and molecular mechanisms. Molecular findings with functional consequences can then be validated in human subjects. This approach may provide a shorter path to a better understanding of and much-needed therapeutic targets for 22q11DS and schizophrenia.
Footnotes
Acknowledgements
We thank Joshua Stokes and Julie Groff for assistance with art work, Dr. Angela McArthur for editing this article, and Drs. Jay Blundon and J.J. Westmoreland for constructive comments.
Authors’ Note
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other granting agencies.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported, in part, by the National Institute of Mental Health of the National Institutes of Health (R01 MH095810, R01 MH097742, R01 MH079079 grants to SSZ and by the American Lebanese Syrian Associated Charities.